Novinky v léčbě nejen dětských hemofiliků
Souhrn:
Komrska V, Zápotocká E. Novinky v léčbě nejen dětských hemofiliků. Remedia 2022; 32: 191–196.
Vrozená hemofilie A či B jsou v současné době dobře kontrolovatelné nemoci, a to pomocí koncentrátů koagulačních faktorů VIII (FVIII) a IX (FIX) vyráběných rekombinantní technologií nebo z lidské plazmy, nověji také s využitím nefaktorové léčby. Tradiční koncentráty dokáží zastavit krvácení a dají se použít i k profylaktické léčbě, jejich společnou nevýhodou je však krátký poločas účinku a aplikace výhradně intravenózní cestou. Výzkum a vývoj léků na hemofilii šel jednak cestou prodloužení poločasu účinku koncentrátů koagulačních faktorů, jednak u nefaktorové léčby využitím jiných principů dosažení očekávané hemostázy, navíc s benefitem odstranění nutnosti intravenózní aplikace a snížením imunogenicity. Souběžně s tím pokračuje intenzivní výzkum a příprava genové léčby, která je již ve stadiu klinických hodnocení u lidí. Moderní léčba hemofilie staví pacienta do centra péče s maximálním důrazem na personalizaci léčby, tedy se zohledněním jeho životního stylu, fyzické aktivity či již přítomného poškození kloubů, u starších hemofiliků rovněž se zohledněním přítomných komorbidit.
Summary:
Komrska V, Zapotocka E. Updates in the treatment of not only paediatric haemophiliacs. Remedia 2022; 32: 191–196.
Congenital haemophilia A and B are currently well‑controllable diseases with the help of coagulation factor VIII and (FVIII) and IX (FIX) concentrates manufactured with recombinant technology or from human plasma, and more recently with the use of non‑factor treatment. Traditional concentrates can stop the bleeding and can be used in prophylactic treatment. However, their common disadvantage is a short half‑life of action and an exclusively intravenous administration. The research and development of drugs aimed to increase the half‑life of action of coagulation factor concentrates and, in the non‑factor treatment, to use alternative principles of achieving haemostasis, with the benefit of removing the barrier of exclusively intravenous administration and lowering the immunogenicity. Simultaneously, intensive research and preparation of gene therapy continues, achieving the level of clinical trials in humans. Modern haemophilia treatment puts the patient into the centre of care, with maximal focus on treatment personalisation, considering their lifestyle, physical activity or already present joint damage and their comorbidities in older patients.
Key words: haemophilia, substitution treatment, factor VIII and IX concentrate, half‑life of action, prolonged action time , bypass drugs, non‑factor treatment, gene therapy, personalisation of treatment.
Hemofilie A a B jsou vrozená
krvácivá onemocnění, jež mohou v nejtěžších formách
vést k významným krvácivým projevům, které při
nedostatečné léčbě mohou být invalidizující až život
ohrožující. Historickým zlomem v péči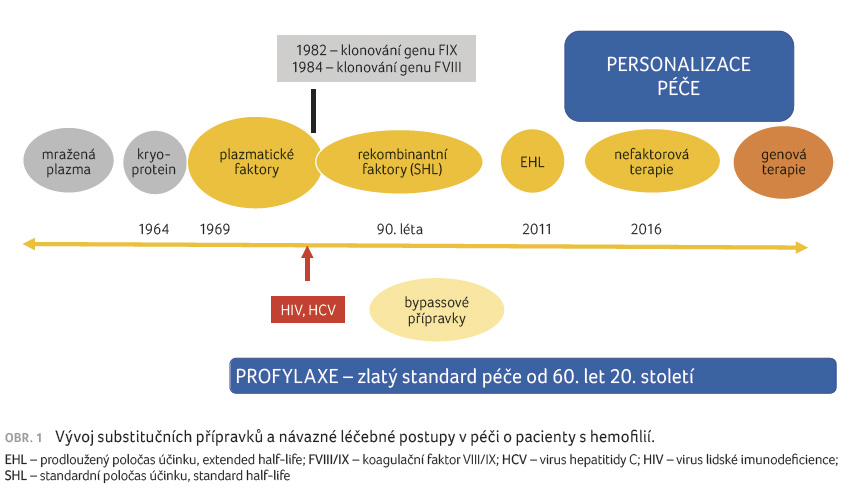 o hemofiliky
byla příprava koncentrovaného faktoru VIII (FVIII) a IX
(FIX), jež umožnily efektivní léčbu krvácení a také
zavedení tzv. profylaktické péče, tedy pravidelného podávání
koncentrátů srážlivých faktorů s cílem
konvertovat těžkou formu hemofilie na středně těžkou,
a tím předcházet řadě spontánních i traumatických
krvácení. Jednadvacáté století přineslo další, až revoluční
léčebné přístupy, a díky novým technologiím umožnilo
efektivnější dialog s nemocnými a současně rovněž
další vylepšení preventivní péče v ambulancích
specialistů na hemofilii [1] (obr. 1).
o hemofiliky
byla příprava koncentrovaného faktoru VIII (FVIII) a IX
(FIX), jež umožnily efektivní léčbu krvácení a také
zavedení tzv. profylaktické péče, tedy pravidelného podávání
koncentrátů srážlivých faktorů s cílem
konvertovat těžkou formu hemofilie na středně těžkou,
a tím předcházet řadě spontánních i traumatických
krvácení. Jednadvacáté století přineslo další, až revoluční
léčebné přístupy, a díky novým technologiím umožnilo
efektivnější dialog s nemocnými a současně rovněž
další vylepšení preventivní péče v ambulancích
specialistů na hemofilii [1] (obr. 1).
Substituční léčba
Současným pilířem léčby hemofilie jsou substituční přípravky, ať již vyrobené z krevní plazmy, nebo rekombinantní technologií, se standardním nebo prodlouženým poločasem účinku [2]. V České republice aktuálně používáme u malé části pacientů koncentráty vysoké čistoty vyráběné z plazmy a je registrována řada rekombinantních přípravků FVIII a FIX.
Koncentráty FVIII vyráběné rekombinantní technologií
V současné době jsou používány dvě genové technologie k výrobě těchto přípravků. První postup spočívá v produkci FVIII spolu s von Willebrandovým faktorem (vWF) na tkáňových kulturách ovariálních buněk čínského křečka a v konečném produktu je vWF odstraněn immunoafinní chromatografií. Při druhém postupu je získáván přímo čistý FVIII na tkáňových kulturách ledvinných buněk křeččích mláďat. Biochemické, farmakokinetické a klinické vlastnosti obou produktů jsou prakticky stejné jako u koncentrátů vyráběných z plazmy, specifická aktivita je však mnohonásobně vyšší a dosahuje více než 5 000 IU (u některých přípravků více než 10 000 IU) na miligram proteinu. Druhá generace rekombinantních přípravků obsahuje zvířecí nebo lidské proteiny v buněčném médiu, ne však již ve finálním produktu. Třetí generace rekombinantních přípravků FVIII již neobsahuje žádné plazmatické proteiny lidské ani zvířecí nejen ve finálním produktu, ale ani v buněčném médiu, produkt je stabilizován složitými glycidy. Tyto přípravky lze z mikrobiologického hlediska označit za první stoprocentně bezpečný FVIII. Možnost přenosu lidské nebo zvířecí infekce je zde zcela vyloučena. V rekombinantní technologii je využívána celá genová informace FVIII či je odstraněna B doména (z angl. B domain deleted, BDD).
Substituční terapie hemofilie B
Podobně jako u FVIII i zde se vývoj ubíral cestou lyofilizovaných koncentrátů. Tyto koncentráty FIX jsou v lyofilizované podobě dlouhodobě stabilní i při pokojové teplotě, rozpouštějí se v malém množství rozpouštědla, takže při jejich použití odpadá objemové přetížení pacienta. Moderní vysoce čištěné koncentráty FIX neobsahují příměs aktivovaných koagulačních faktorů a jejich trombogennost je velmi nízká. Na trhu a v běžném klinickém použití jsou rovněž k dispozici rekombinantní koncentráty FIX třetí generace.
Mikrobiologická bezbečnost a imunogenicita
Vývoj substitučních přípravků se sníženou imunogenicitou je zaměřen na redukci počtu pacientů s inhibitorem (protilátka proti FVIII/FIX). Inhibitor je v současné době nejzávažnější komplikací hemofilie, která postihuje asi 30 % pacientů s těžkou hemofilií A a 3 % pacientů s těžkou hemofilií B. Přítomnost inhibitoru znemožňuje léčbu běžnými koncentráty, které jsou protilátkou neutralizovány, takže je tato terapie méně účinná a extrémně nákladná.
Bezpečnost substitučních přípravků zohledňuje mikrobiologickou bezpečnost a imunogenicitu. Z hlediska mikrobiologické bezpečnosti představují vrchol nabídky rekombinantní přípravky, z hlediska schopnosti indukovat tvorbu inhibitoru probíhá široká diskuse, zda nejsou rekombinantní přípravky více imunogenní než přípravky vyráběné z plazmy.
Substituční přípravky s prodlouženým poločasem
Zásadní nevýhodou substitučních přípravků užívaných klasicky do druhé dekády 21. století (tzv. produkty o standardním biologickém poločasu − standard half-life, SHL) je jejich krátký poločas účinku − u FVIII 8−12 hodin, u FIX 18−24 hodin. Poslední dva až tři roky jsou v ČR k dispozici také koncentráty FVIII/FIX s prodlouženou dobou účinku (extended half-life, EHL). Prodloužení biologického poločasu lze dosáhnout několika způsoby [3]:
- Fúze Fc – fúze biologicky aktivního proteinu s Fc doménou lidského imunoglobulinu G1 (IgG1) je spojena s ochranou proteinu před lysozomální degradací a prodloužením jeho biologického účinku. Rekombinantně vyrobený FVIII zbavený B domény je kovalentní vazbou navázán na Fc doménu lidského IgG1. Ve studiích s těmito přípravky bylo prokázáno 1,6−1,7násobné prodloužení času eliminace a poločasu FVIIIFc až na 19 hodin. Podobně ošetřená přirozená molekula FIX vykazuje více než trojnásobné prodloužení doby účinku a prodloužení poločasu až na 82 hodin.
- Fúze s albuminem − albumin je v krvi přirozeně se vyskytující protein a jeho biologický poločas zde dosahuje více než 20 dnů. Je široce používán jako bílkovinný stabilizátor při výrobě řady léčiv. Rekombinantní FIX s navázaným rekombinantním albuminem (rIX FP) dosahuje poločasu přibližně 4,3krát delšího než u běžných přípravků FIX.
- Pegylace znamená kovalentní připojení polyetylenglykolu (PEG) k proteinu, peptidu nebo malé lékové molekule. Pegylace prodlužuje biologický poločas bílkovin několika mechanismy: a) zvětšením jejich molekuly, b) snížením jejich citlivosti k proteolytickému štěpení a degradaci a c) změnou vlastností povrchového elektrického náboje dochází k interferenci s procesy clearance. Pegylací ošetřený FVIII má ve srovnání s běžnými přípravky taktéž zhruba 1,8násobně prodloužený biologický poločas. Pegylovaný FIX má poločas prodloužen oproti běžnému FIX přibližně pětkrát. V současnosti je v České republice léčba koncentráty FVIII či FIX s navázaným PEG povolena Státním ústavem pro kontrolu léčiv (SÚKL) pouze u pacientů starších 12 let.
- Polysialyzace − vazba koagulačního proteinu na kyselinu polysialovou – vede k podobnému efektu jako pegylace a prodlužuje poločas účinku FVIII/FIX.
- Jednořetězcový rekombinantní FVIII − rFVIII se skládá ze dvou spojených proteinových řetězců, lehkého a těžkého, které lze různými způsoby oddělit. Lze však připravit jednořetězcový rFVIII, kde jsou spojeny lehký a těžký řetězec kovalentní vazbou. Takto připravený jednořetězcový faktor vykazuje zvýšenou molekulární integritu i stabilitu a má zvýšenou afinitu k vWF, což může vést k jeho snížené imunogenicitě.
Příchod EHL produktů umožnil řadě pacientů snížit frekvenci profylaktického podávání FVIII (typicky nyní každý 3. den či dvakrát týdně, u některých pacientů i 1× za 5 dnů) při zachování stejné ochrany jako při frekventnější profylaxi se SHL produkty. U jiných nemocných se dá využít efektu dosažení vyšších minimálních (trough) koncentrací, jež jsou pro řadu aktivit výhodnější, při zachování stejné frekvence aplikací jako při léčbě SHL přípravky (graf 1A, B) [4]. V případě EHL produktů FIX bylo možno dosáhnout frekvence aplikace 1× za 7−10 dnů (v některých případech i 14 dnů, popř. méně) v závislosti na krvácivých projevech. Na okraj je nutno podotknout, že m
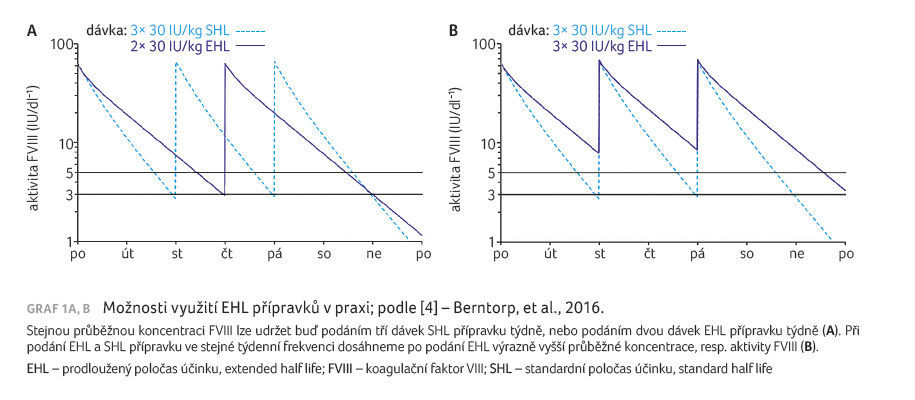
odifikované EHL produkty mají svá laboratorní specifika, pokud jde o stanovení koncentrací faktorů, a tato specifika (výběr reagens pro testy) je nutno zohlednit [5].
Nefaktorová léčba
Nefaktorová léčba představuje zcela nový přístup k profylaxi u hemofilie. Efektivní hemostázy je dosaženo jiným způsobem než podáváním koncentrátů deficitních faktorů [6].
- Bispecifická
protilátka emicizumab je namířena proti FIXa na jedné
straně a proti FX na straně druhé (obr. 2)
a jejich vazbou dojde k prostorovému sblížení obou
faktorů s následnou aktivací FX na FXa. Bispecifická
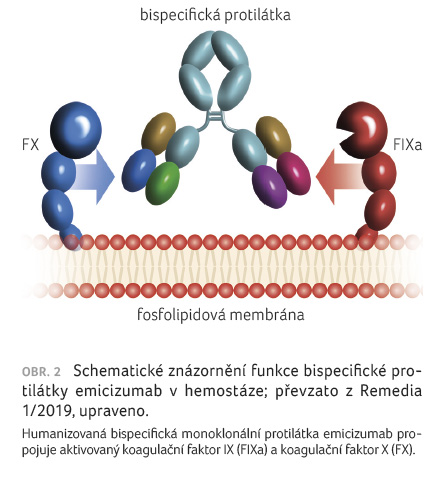 protilátka zde nahrazuje (mimikuje) funkci FVIII jako kofaktoru.
Její biologický poločas činí 2−3 týdny, aplikuje se podkožně
a efektivně funguje u hemofilie A s inhibitorem
i bez inhibitoru [7]. Data z klinických studií ukázala
významné snížení počtu léčených krvácivých epizod
ve srovnání s předchozí profylaxí koncentráty
srážlivých faktorů. Emicizumab je již v běžném klinickém
použití a také data z reálné praxe potvrzují velmi
dobrý potenciál prevence krvácení, avšak v případě
některých operačních výkonů či většího krvácení je
potřeba podat jednorázově či krátkodobě koncentrát FVIII nebo
u nemocných s inhibitorem tzv. bypassový lék (aktivovaný
protrombinový komplex [aPCC], rFVIIa − viz dále).
Dávkování emicizumabu se neliší napříč věkovými
kategoriemi, je možno jej podávat 1× za 7 či 14 dnů,
popřípadě 1× za 28 dnů. Jeho efekt přetrvává v těle
až šest měsíců po vysazení. Emicizumab významně
ovlivňuje některé laboratorní testy, což je také nutno vést
v patrnosti. Emicizumab se stává standardem přístupu
k profylaxi u pacientů s inhibitorem FVIII.
protilátka zde nahrazuje (mimikuje) funkci FVIII jako kofaktoru.
Její biologický poločas činí 2−3 týdny, aplikuje se podkožně
a efektivně funguje u hemofilie A s inhibitorem
i bez inhibitoru [7]. Data z klinických studií ukázala
významné snížení počtu léčených krvácivých epizod
ve srovnání s předchozí profylaxí koncentráty
srážlivých faktorů. Emicizumab je již v běžném klinickém
použití a také data z reálné praxe potvrzují velmi
dobrý potenciál prevence krvácení, avšak v případě
některých operačních výkonů či většího krvácení je
potřeba podat jednorázově či krátkodobě koncentrát FVIII nebo
u nemocných s inhibitorem tzv. bypassový lék (aktivovaný
protrombinový komplex [aPCC], rFVIIa − viz dále).
Dávkování emicizumabu se neliší napříč věkovými
kategoriemi, je možno jej podávat 1× za 7 či 14 dnů,
popřípadě 1× za 28 dnů. Jeho efekt přetrvává v těle
až šest měsíců po vysazení. Emicizumab významně
ovlivňuje některé laboratorní testy, což je také nutno vést
v patrnosti. Emicizumab se stává standardem přístupu
k profylaxi u pacientů s inhibitorem FVIII. - Inhibice přirozených antikoagulancií při deficitu FVIII či FIX navozuje novou rovnováhu koagulačního systému a vede ke snížení tendence ke krvácení. Jedná se o látky zatím v klinickém vývoji − např. o inhibiční protilátku proti inhibitoru tkáňového faktoru (tissue factor pathway inhibitor, TFPI) concizumab. TFPI je inhibitor v plazmě, který moduluje koagulaci spuštěnou tkáňovým faktorem (TF). TFPI váže a inhibuje komplexy TF FVII/FVIIa. Pokud je TFPI inhibován, zvyšuje se generace trombinu. TFPI lze inhibovat i aptamery, nicméně jejich klinické využití je zatím zkoumáno. Jinou cestu představuje redukce funkce antitrombinu pomocí interferujících malých RNA molekul, konkrétně studovaných pod názvem fitusiran. Jedná se o zajímavé látky, které mají potenciál ovlivnit management jak hemofilie A, tak i hemofilie B, a pravděpodobně také dalších vrozených krvácivých onemocnění.
Další možnosti léčby
K zajištění zejména případů krvácení a invazivních výkonů u pacientů s inhibitorem se používají tzv. bypassové přípravky, které aktivují FX na Xa i bez přítomnosti FVIII/FIX. V klinickém použití jsou v nezměněné podobě již řadu let přípravky aPCC a rFVIIa. Ani v této oblasti se však vývoj nezastavil a jsou vyvíjeny nové molekuly rFVIIa, které by měly mít lepší farmakologické vlastnosti, zvláště ve smyslu prodloužení účinku, jenž je u stávajícího rFVIIa velmi krátký. K prodloužení efektu a k výraznější generaci trombinu vede vazba s pegylovanými lipozomy, k rychlejšímu a déletrvajícímu účinku vede analog rFVIIa vatreptacog alfa. Zkoumány jsou rovněž varianty rFVIIa vzniklé různými arteficiálními změnami aminokyselin v řetězci rFVIIa.
Vepřový FVIII představuje staronový prostředek pro léčbu hemofiliků s vysokým titrem inhibitoru a pacientů se získanou hemofilií. Plazmatický vepřový FVIII byl s úspěchem používán již před několika desítkami let. Využívalo se nízké zkřížené reaktivity s protilátkami a lepší klinické odpovědi při léčbě tímto přípravkem v porovnání s léčbou bypassovými přípravky. Výhodou byla rovněž možnost stanovení koncentrace porcinního FVIII a monitorace léčby, zatímco u bypassových přípravků je laboratorní sledování terapie obtížné. V roce 2004 byl přípravek stažen z trhu z důvodu přenosu prasečího parvoviru. Vývoj vepřového rFVIII tento problém vyřešil a přípravek je již k dispozici na trhu.
Genová terapie
Naději pro všechny pacienty trpící hemofilií představuje do budoucna genová terapie [8]. Odhalení struktury a klonování genu pro FVIII a FIX na počátku 80. let minulého století umožnilo nejen výrobu rekombinantních substitučních přípravků, ale dalo rovněž základní teoretický předpoklad pro tuto terapii. Hemofilie se svou jednoduchou dědičností a snadným sledováním účinku léčby (koncentrace FVIII/FIX) je ideálním modelem pro genovou terapii. Po počátečním entuziasmu se ukázalo, že genová terapie má řadu úskalí a její zavedení nebude tak rychlé, jak se předpokládalo. Po celé řadě experimentálních prací na zvířatech byly již v roce 1999 zahájeny ve Spojených státech amerických a později v dalších zemích první klinické zkoušky u lidí. Jako schůdnější se ukázala genová léčba hemofilie B a v současné době lze shrnout, že dospělý nemocný s těžkou hemofilií B, který je HCV (virus hepatitidy C) RNA negativní a nemá protilátky proti adeno asociovanému viru (AAV), vektoru přenosu, může podstoupit intravenózní infuzi vektoru AAV s genem přirozeného FIX. Po infuzi lze očekávat nejméně tříletou sekreci FIX s dosažením koncentrace kolem 5 %. Jedná se o výrazný úspěch, kdy těžká forma hemofilie přechází v lehkou a dramaticky se mění klinický průběh nemoci. Zajímavá je varianta FIX Padua, která vede k ještě vyšším dosaženým plazmatickým koncentracím FIX. V běhu je ještě několik dalších klinických studií u hemofilie B, kde lze očekávat podobné výsledky.
Genová léčba hemofilie A je obtížnější, což je dáno především větší velikostí genu pro FVIII a jeho obtížnější inkorporací do hostitelské buňky. Probíhající studie ukazují po převodu vektoru taktéž u většiny osob dosažení koncentrací FVIII v pásmu lehké hemofilie či normy, problémem se zatím zdá dlouhodobé udržení exprese transgenu. Genová terapie postihuje somatické buňky a může zlepšit zdravotní stav, nebo i vyléčit daného jedince. Protože ale nepostihuje zárodečné buňky, nezabrání přenosu defektního genu na pacientovy dcery, které se tak − stejně jako dosud − stanou přenašečkami. O skutečném vyléčení hemofilie budeme moci hovořit teprve tehdy, až bude k dispozici genová léčba postihující i zárodečné buňky.
Přístup k pacientům
s hemofilií ve 21. století
Moderní léčba hemofilie staví
pacienta do centra multidisciplinární péče, přičemž
systém komplexních center (comprehensive care centre, CCC)
a menších léčebných center (hemophilia treatment centre,
HTC) umožňuje dobrou spolupráci a zajištění optimální
péče, včetně péče multioborové. Profylaxe primární
a sekundární je u hemofiliků s krvácivými projevy
dnes již standardem péče, významně narůstá podíl nemocných
s terciární profylaxí [9,10]. Vedle dostupnosti nových léků
a jejich zavádění do klinické praxe je zásadní jejich
efektivní podávání a vhodná indikace. V případě
profylaxe koncentráty FVIII/FIX se v současnosti často léčba
opírá také o farmakokinetické vyšetření (PK profil), jež
je díky některým dostupným bayesiánským modelům (např.
akademický projekt WAPPs HEMO) dobře proveditelné i při
omezeném množství odebraných vzorků [9]. Cílem je nastavit
profylaxi tak, aby co nejlépe vyhovovala životnímu stylu daného
jedince či míře poškození jeho kloubů, popřípadě již
existujícím přidruženým onemocněním (graf 2). V dialogu
s pacienty mohou pomoci aplikace do chytrých telefonů,
jež využívají PK profil, zlepšují edukaci hemofiliků a jejich
rodin a současně zvyšují v řadě případů adherenci
k léčbě. Detekci časného, zatím i subklinického
poškození kloubů pomáhá bedside ultrazvukové vyšetření
v rukou hematologa či ortopeda, které je v ČR na řadě
pracovišť již také k dispozici [11].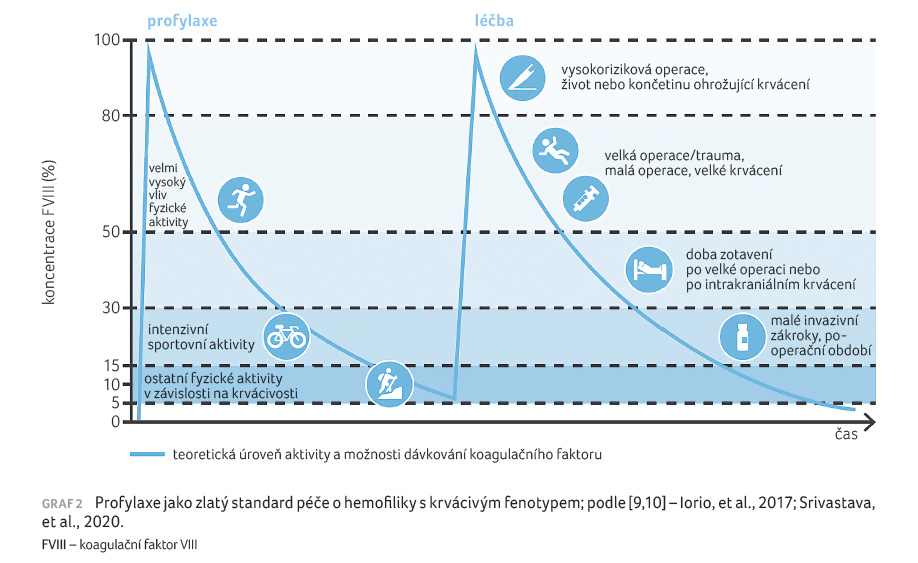
Všechny pokroky a nové trendy v přístupu k hemofilii rovněž reflektují péči o přenašečky, zajištění prenatální a postnatální péče o děti s hemofilií, časné stanovení diagnózy hemofilie, edukaci rodin a zahájení efektivní profylaxe u řady dětí v útlém věku. Na druhé straně pak stojí stárnoucí populace hemofiliků a hledání optimálního nastavení jejich léčby ve vztahu k chorobám spojeným se stárnutím [10].
Třetí vydání doporučení Světové hemofilické federace (World Federation of Hemophilia, WFH) z roku 2020 navrhuje novou definici profylaxe jako pravidelného podávání hemostatické či jiné látky s cílem prevence krvácení hemofiliků s možností pomoci vést aktivní život a dosáhnout kvality života srovnatelné s lidmi bez hemofilie. Zda toto bude možnou realitou pro většinu hemofiliků, ukáže blízká budoucnost.
Seznam použité literatury
- [1] Blanchette VS, Key NS, Ljung LR, et al. Definitions in hemophilia: communication from the SSC of the ISTH. J Thromb Haemost 2014; 12: 1935−1939.
- [2] Oldenburg J. Optimal treatment strategies for hemophilia: achievement and limitations of current prophylactic regimens. Blood 2015; 125: 2038−2044.
- [3] Mannucci PM. Haemophilia therapy: the future has begun. Hematologica 2020; 105: 545−553.
- [4] Berntorp E, Negrier C, Gozzi P, et al. Dosing regimens, FVIII levels and estimated haemostatic protection with special focus on rFVIIIFc. Haemophilia 2016; 22: 389−396.
- [5] Collins P, Chalmer E, Chowdary P, et al. The use of enhanced halflife coagulation factor concentrates in routine clinical practice: guidance from UKHCDO. Haemophilia 2016; 22: 487−497.
- [6] Massimo F, Mannucci PM. Non‑factor replacement therapy for haemophilia: a current update. Blood Transfus 2018; 16: 457–461.
- [7] Franchini M, Marano G, Pato I, et al. Emicizuman for the treatment of haemophila A: a narrative review. Blood Transfus 2019; 17: 223−228.
- [8] Batty P, Lillicrap D. Advances and challenges for haemophilia gene therapy. Hum Mol Genet 2019; 28: R95−101.
- [9] Iorio A, Blanchette V, Blatny J, et al. Estimating and interpreting the pharmacokinetic profiles of individual patients with hemophilia A or B using a population pharmacokinetic approach: communication from the SSC of the ISTH. J Thromb Haemost 2017; 15: 2461−2465.
- [10] Srivastava A, Santagostino E, Dougall A, et al. WFH Guidelines for the management of hemophilia, 3rd edition. Haemophilia 2020; (Suppl 6): 1−158.
- [11] Rodrigues C, Rodriguez‑Merchan EC, Alvarez‑R MT, et al. The value of HEAD‑US system in detecting subclinical abnormalities in joints of patients with hemophilia. Expert Rev Hematol 2018; 11: 253−261.