Autoimunitní hepatitis
Autoimunitní hepatitis (AIH) je autoimunitní zánět jaterní tkáně. Je charakterizován přítomností interface hepatitidy v histologickém obraze, hypergamaglobulinémií a přítomností autoprotilátek v séru. Diagnóza vyžaduje vyloučení ostatních podobných jaterních lézí – Wilsonovy choroby, chronické virové hepatitidy, α-1-antitrypsinové deficience, nealkoholické steatohepatitidy, primární biliární cirhózy (PBC), primární sklerózující cholangoitidy (PSC) a autoimunitní cholangoitidy. V léčbě je používán buď samotný prednison nebo prednison v kombinaci s azathioprinem.
Skórovací kritéria
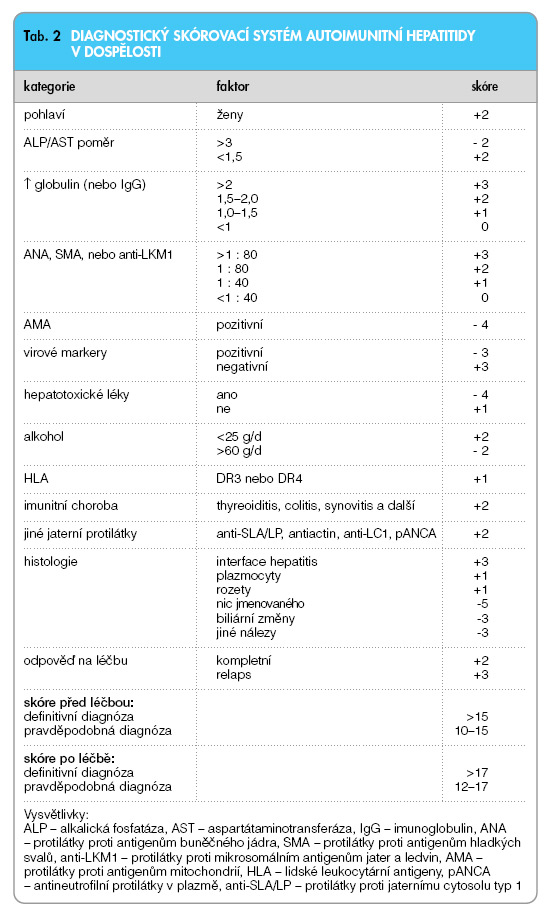
Kritéria kodifikoval panel mezinárodních expertů v roce 1992 s následnou úpravou v roce 1999 [1]. Původní požadavek 6měsíčního trvání choroby byl opuštěn. Imunosérologické testy nutné k diagnóze zahrnují antinukleární protilátky (ANA), protilátky proti hladkému svalu (ASMA) a protilátky proti liver/kidney mikrosomu type 1 (anti-LKM1). U typu 1 je častá přítomnost antineutrofilních protilátek v cytoplazmě (pANCA) [2]. Jednoznačná diagnóza AIH vyžaduje 1) vyloučení ostatních podobných nemocí, 2) laboratorní nález s vyjádřenou imunoreaktivitou a 3) histologický nález interface hepatitidy. Pravděpodobnou diagnózu označíme při nálezu kompatibilním s AIH, ale nedostatečném pro jednoznačnou diagnózu. Nemocní nemají konvenční protilátky, ale mohou být pozitivní na jiné protilátky (např. anti-ASGPR, anti-SLA/LP, antiactin, anti-LC1). Systém navržený mezinárodní skupinou popisuje různé manifestace AIH a slouží ve svém celkovém skóre k popisu tíže onemocnění. Lze jej také využít k hodnocení úspěšnosti léčby. Má také význam při výzkumu AIH (tab. 2).
Patogeneze AIH
Přesný mechanismus vzniku choroby není znám. Mezi nejoblíbenější hypotézy patří přítomnost různých interaktivních faktorů – spouštěcího agens, genetických predispozic, různých poruch nabídky antigenu, aktivace imunokompetentních buněk, expanze efektorových buněk apod. Mezi možné spouštěcí faktory zahrnujeme infekční agens, léky a toxiny. Interval od působení agens k vzniku choroby může být různě dlouhý a spouštěcí faktor nemusí být přítomen k udržování choroby. Ke ztrátě tolerance k vlastní tkáni mohou vést molekulární mimikry cizího nebo vlastního antigenu. Ke genetickým predispozicím řadíme změny v MHC II (hlavní histokompatibilní komplex) s určitou predispozicí různých etnických skupin. Destrukce jater je způsobena buď buňkami zprostředkovanou cytotoxicitou nebo cytotoxicitou závislou na protilátkách nebo obojím mechanismem. Buněčná cytotoxicita závisí na klonální expanzi CD8+ T buněk za pomoci lymfokinů typu 1. Tyto projevy mohou být ovlivněny genetickými polymorfismy takových cytokinů, jako např. TNF-α. Cytotoxicita zprostředkovaná protilátkami je naopak ovlivněna cytokiny 2. typu a destrukcí hepatocytů NK buňkami. Tento mechanismus závisí na diferenciaci CD4+ buněk, která je opět ovlivněna polymorfismy cytokinů.
Klasifikace
Na základě imunosérologických markerů byly navrženy 3 typy AIH [3]. Pouze dva z nich mají téměř vzájemně se vylučující autoprotilátky a žádný nemá izolovanou příčinu nebo typický klinický fenotyp. Mezinárodní skupina expertů (International Autoimmune Hepatitis Group) nepodpořila subklasifikaci AIH, která má v podstatě pouze popisnou roli, i když v praxi je stále možné se s tímto dělením setkat (tab. 1).
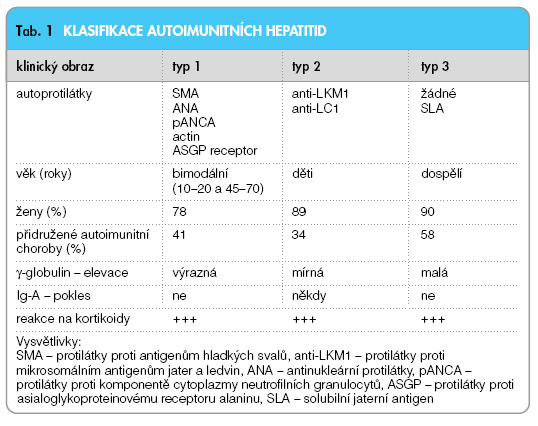
Typ 1 mívají častěji ženy (78 %) a 41 % z nich má přidruženou jinou autoimunitní chorobu (zejména thyreopatii). Typ 1 má akutní začátek častěji. Typ 2 postihuje více děti, typ 3 je klinicky neodlišitelný od typu 1. Existují tzv. variantní formy – mají autoimunitní rysy, ale nesplňují kritéria. Mají projevy AIH, a navíc ještě symptomatologii jiného chronického onemocnění jater – primární biliární cirhózy nebo sklerózující cholangoitidy. autoimunitní cholangoitida má rysy AIH a primární biliární cirhózy (PBC) bez antimitochondriálních protilátek (tzv AMA negativní PBC) nebo primární sklerózující cholangoitidy (PSC) malých duktů.
Prevalence, prognostická kritéria a příznaky
V Evropě je prevalence okolo 1,9 případů na 100 000 osob za rok. Prognóza je ovlivněna mírou zánětu při iniciálním vyšetření, tj. elevace AST, GMT, a mírou postižení v histologickém nálezu. Ke spontánnímu ústupu dochází v 13–20 %. Nejčastějším příznakem je únava, ikterus a tlaky v epigastriu. Objektivně nalézáme vedle ikteru hepatomegalii. V laboratoři pak elevaci jaterních transamináz a polyklonální hypergamaglobulinémii.
Léčba
V léčbě používáme buď samotný prednison nebo prednison v kombinaci s azathioprinem (AZA). Uspokojivou odpověď na léčbu nelze předpovídat [4]. Vzhledem k nežádoucím účinkům dlouhodobé léčby prednisonem se více doporučuje kombinovaná léčba s AZA. U žen v postmenopauze je nutné zabránit kostním nežádoucím projevům – iniciovat pravidelný program cvičení, podávání vitaminu D a kalcia. Vhodné je zvážit u nemocných s osteopenií podání bisfosfonátů (alendronat 10 mg/den nebo etidronat 400 mg/den vždy 2 týdny každé 3 měsíce). U žen, které by plánovaly těhotenství, je žádoucí vyhnout se AZA. Cílem léčby je dosažení remise, což znamená nepřítomnost symptomů (ústup známek zánětu, tj. AST <2x norma) a nepřítomnost histologické aktivity (obvykle 3–6 měsíců po ústupu laboratorní aktivity). Selhání léčby znamená zhoršení i během léčby – zvýšení hladin AST nebo hladiny bilirubinu, vznik ascitu, encefalopatie apod. Přerušíme pak konvenční léčbu kortikoidy a nasadíme vysoké dávky. Inkompletní odpověď nesplňuje kritéria remise, nepřítomnost remise do 3 let od zahájení znamená, že dosažení remise je nepravděpodobné. Toxické projevy léků vedou k předčasnému vysazení nebo k redukci dávky. Prednison samotný nebo v kombinaci s AZA vede k remisi (klinické, biochemické a histologické) v 65 % do 3 let. Průměrná doba léčby je 22 měsíců. Léčba prokazatelně prodlužuje život. Nemocní, kteří mají již cirhózu, by měli být léčeni stejně. Pacienti, kteří vstoupí do remise, mají často relaps po vysazení léků (50 % do 6 měsíců po vysazení nebo 70–86 % do 3 let). Znovu zahájíme léčbu s novou remisí. Pacienti, kteří měli alespoň 2 relapsy, vyžadují kontinuálníléčbu, 87 % z nich stačí menší dávka prednisonu denně (v průměru 7,5 mg/den). Dávka se vytitruje na nejnižší možnou, která ještě zabrání symptomatologii a udrží sérové aminotransferázy <5x velikost normy. Alternativní strategií je podávání 2 mg/kg/den AZA (nemocní bez těžké cytopenie a těhotenství). Oba režimy nebyly proti sobě posuzovány, ale není důvod preferovat jeden oproti druhému, u obou se předpokládá stejná účinnost. Dojde-li k selhání léčby, nasazujeme vyšší dávky prednisonu (60 mg/den) nebo kombinaci 30 mg prednisonu se 150 mg AZA denně. Úspěšnost klinická a biochemická je v cca 70 % do 2 roků, histologická ale jenom ve 20 %. Vždy je třeba při prvních známkách dekompenzace jater zvažovat transplantaci. Alternativní režimy zahrnují ciclosporin, mycophenolat mofetil, ursodeoxycholovou kyselinu, budesonid, 6-merkaptopurin, methotrexát a cyclophosphamid. Tato léčba patří ale na specializované pracoviště. U nekompletní remise doporučujeme pokračovat v podávání nízké dávky prednisonu.
Seznam použité literatury
- [1] Alvarez F, Berg P, Bianchi F, et al. International Autoimmune hepatitis group report: Review of criteria for diagnosis of autoimmune hepatitis. J Hepatol 1999;31:929.
- [2] Vergani D, Alvarez F, Bianchi F, et al Liver autoimmune serology: a consensus statement from the committee for autoimmune serology of the International Autoimmune Hepatitis Group. J Hepatol 2004;41:677–83.
- [3] Ben-Ari Z, Czaja A. Autoimmune haptitis and its variant syndromes. Gut 2001;49:589–94.
- [4] Czaja A. Autoimmune hepatitis. In: Slezinger and Fordtran´s Gastrointestinal and Liver Disease, 7 th ed. Saunders 2002:1462–73.