Diagnostika lysozomálních střádavých onemocnění
Souhrn:
Ješina P. Diagnostika lysozomálních střádavých onemocnění. Remedia 2019; 29: 82–91.
Lysozomální střádavá onemocnění (lysosomal storage disorders, LSD) představují velmi širokou skupinu dědičných metabolických poruch, která zahrnuje přibližně 60 nemocí. Základní patofyziologickou podstatou nemocí je porucha metabolismu složených sacharidů, lipidů nebo proteinů v lysozomech, což vede k jejich hromadění. V tomto článku uvádíme základní přehled LSD, jejich klinické projevy a možnosti současné diagnostiky.
Summary:
Jesina P. Diagnostics of lysosomal storage disorders. Remedia 2019; 29: 82–91.
Lysosomal storage disorders (LSD) present a large group of inborn errors of metabolism with more than 60 diseases. They affect the lysosomal degradation of complex carbohydrates, lipids, and proteins, leading to accumulation in tissues. We summarize LSD, natural course and diagnostics approaches.
Key words: lysosomal storage disorders, diagnosis, enzymology, molecular genetics.
Úvod
Lysozomální střádavá onemocnění (lysosomal storage disorders, LSD) představují velmi širokou skupinu dědičných metabolických poruch zahrnujících přibližně 60 nemocí. Jejich základní patofyziologickou podstatou je porucha metabolismu složených sacharidů, lipidů nebo proteinů v lysozomech, jež vede k jejich hromadění. Lysozomální choroby jsou způsobeny různými poruchami:
- enzymů (např. sfingolipidózy, glykoproteinózy, mukopolysacharidózy),
- aktivátorů enzymů (např. saposinové deficity),
- posttranslační modifikace enzymů (např. mukolipidóza typu II, polysulfatázový deficit),
- funkce protektivních proteinů (např. galaktosialidóza),
- transportních proteinů v lysozomální membráně (např. mukolipidóza typu IV, Niemannova‒Pickova choroba typu C).
Detailní přehled známých LSD
ukazuje tabulka 1, kde
jsou jednotlivé nemoci rozděleny podle patobiochemických skupin.
U každého onemocnění je uveden gen, který kóduje
specifický protein postižený u dané choroby. U nejčastějších
nemocí je vyčíslena prevalence v České republice, kdy data
vycházejí z počtu pacientů diagnostikovaných v Ústavu
dědičných metabolických poruch 1. LF UK a VFN
v letech 1975‒2008. Celková incidence všech LSD je
odhadována na 1 : 5 000‒8 000, což je řadí vedle
mitochondriálních poruch mezi jedny z nejčastějších
dědičných metabolických poruch [1]. Dědičnost většiny LSD je
autozomálně recesivní s některými výjimkami – např.
NCL4 Kufsova choroba (autozomálně dominantní), X vázanou
dědičnost nacházíme u mukopolysacharidózy typu II
(Hunterova syndromu), Fabryho a Danonovy choroby.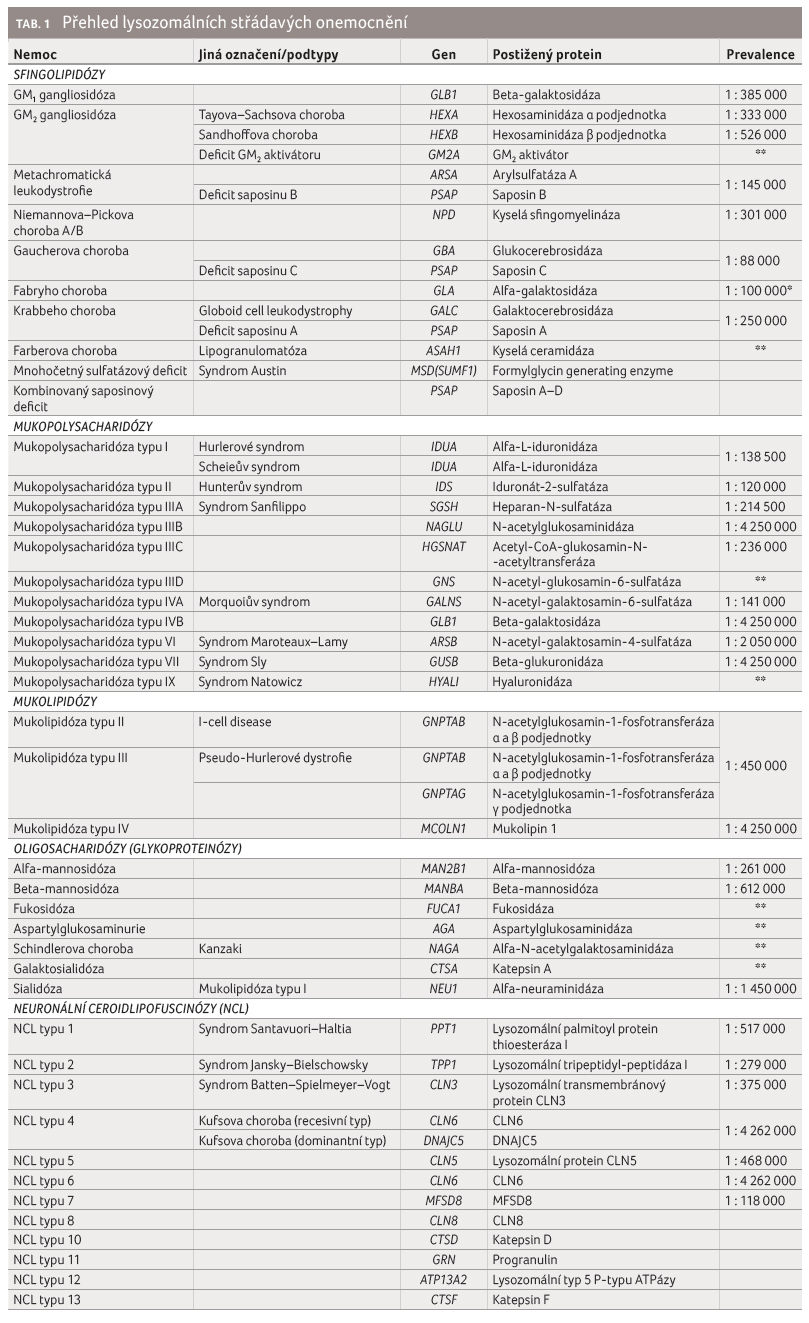
Klinické příznaky lysozomálních
střádavých onemocnění
Spektrum klinických příznaků LSD je i s ohledem na jejich velký počet velmi různorodé. Mezi nejčastější symptomy patří neurologické postižení s psychomotorickou retardací nebo s mentálním deficitem. Často se setkáváme s kraniofaciální dysmorfií, organomegalií (především hepato/splenomegalií) a se skeletálním postižením. Tabulka 2 uvádí souhrnný přehled typických klinických příznaků.
Mukopolysacharidózy (MPS) jsou charakterizovány jako chronické progresivní onemocnění způsobené poruchou funkce enzymů podílejících se na odbourávání glykosaminoglykanů (GAG), což vede k jejich hromadění v lysozomech buněk
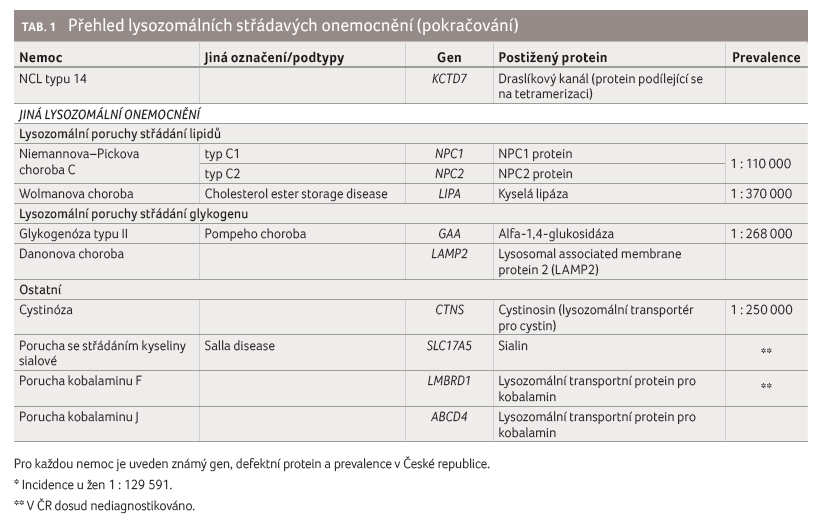
a v extracelulární matrix.
Následně dochází k morfologickým i funkčním změnám
tkání, které se projevují velmi širokou škálou obtíží
a jejich různou tíží (tab. 2). Pro MPS typu I, II, VI a VII je charakteristické
kostní postižení charakteru dysostosis multiplex, organomegalie,
typická kraniofaciální dysmorfie s hrubými rysy
a u některých typů zákal rohovky a neurologické
postižení s psychomotorickou retardací. U MPS typu IVA
nacházíme primárně závažné postižení skeletu bez
neurologického a mentálního postižení. Naopak pro MPS typu
III je typické neurologické postižení a psychomotorická
retardace s regresem vývoje a poruchami chování. Nyní
je popsáno 11 genů, které kódují 11 různých enzymů
podílejících se na metabolismu gaG (tab. 1).
Klinicky se dělí do sedmi typů a u některých jsou
popsány podtypy podle tíže a věku nástupu příznaků,
např. MPS typu I ‒ syndrom Hurlerové (těžší forma
MPS typu I), intermediární forma Hurlerové‒Scheieova syndromu
a Scheieův syndrom (lehčí forma MPS typu I).
Mukopolysacharidóza typu III se dělí na čtyři podtypy podle
postiženého enzymu, 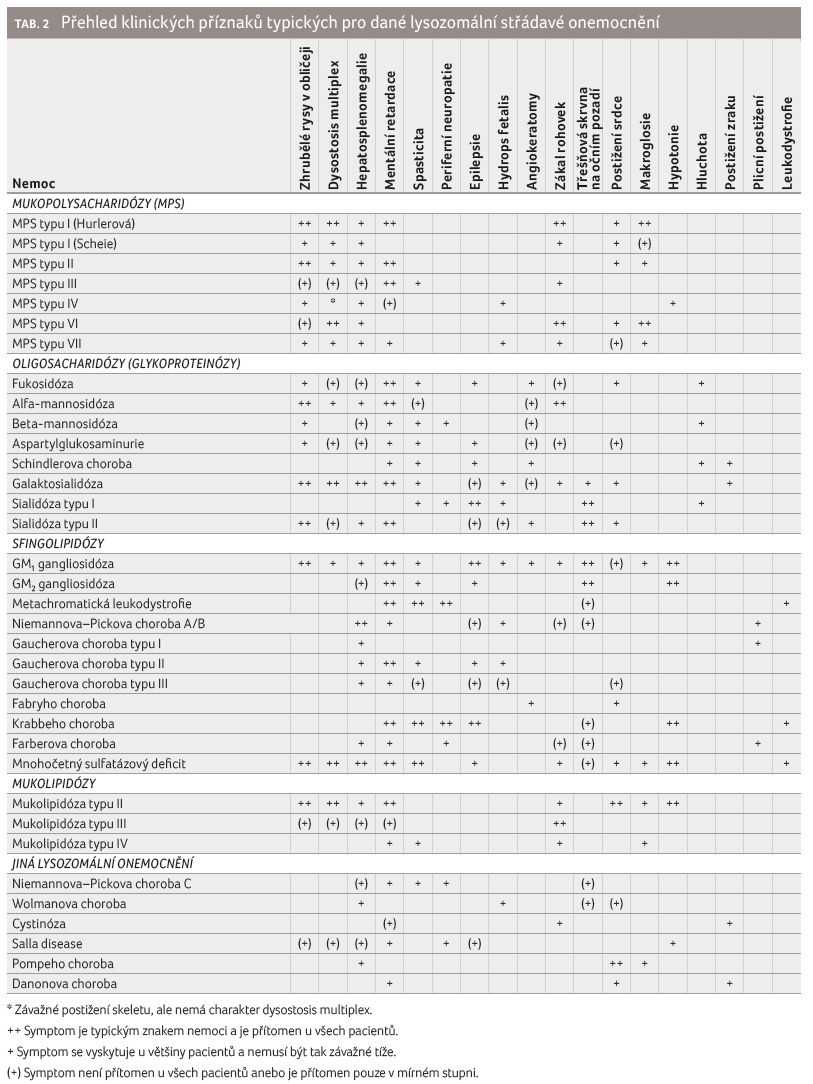 přičemž jednotlivé podtypy mají velmi
podobnou symptomatologii [2,3].
přičemž jednotlivé podtypy mají velmi
podobnou symptomatologii [2,3].
Oligosacharidózy označované také jako glykoproteinózy zahrnují nemoci, u nichž se projevuje postižení funkce lysozomálních enzymů podílejících se na odbourávání glykoproteinů (jejich sacharidové složky) anebo na transportu přes membránu lysozomu. Nejčastější je alfa mannosidóza, pro niž jsou typické příznaky podobné těžší formě MPS typu I (syndrom Hurlerové), kostní změny, postižení sluchu a neurologické příznaky. U beta mannosidózy nacházíme často postižení sluchu, kraniofaciální dysmorfii, mentální postižení a neurologické příznaky. Ostatní glykoproteinózy jsou velmi vzácné [3,4].
Mukolipidózy jsou méně častou skupinou onemocnění, u nichž je defekt v posttranslační glykosylaci lysozomálních hydroláz, což vede k jejich chybnému transportu do buněk a k následnému hromadění v extracelulárním prostoru. U mukolipidózy typu IV je postižen membránový protein mukolipin 1, který je zapojen do transportu lipidů a proteinů. Označení mukolipidóza shrnuje kombinaci příznaků u MPS a sfingolipidóz současně – fenotyp podobný MPS typu I (syndromu Hurlerové) s kraniofaciální dysmorfií, kostními změnami, kardiomyopatií, neurologickým postižením a opožděním psychomotorického vývoje. Pro mukolipidózu typu IV je běžné progresivní postižení zraku se zákalem rohovky a s retinální degenerací a dále achlorhydrie s elevací gastrinu [2,3].
Neuronální ceroidlipofuscinózy (NCL) představují velmi početnou skupinu neurodegenerativních onemocnění, u nichž se v různých tkáních hromadí lipopigment lipofuscin. Pro NCL je charakteristický postupný rozvoj psychomotorické retardace, křečí, ztráty zraku a velmi časně dochází k úmrtí. Klinicky se rozlišují čtyři základní formy podle věku, v němž dochází k nástupu obtíží – infantilní, pozdně infantilní, juvenilní a adultní forma. V souvislosti s rozvojem genetiky došlo k objevu velkého množství genů souvisejících s NCL, a tím se výrazně rozšířilo spektrum typů NCL. Klasická infantilní forma je nejčastěji spojována s NCL1 a s velmi vzácnými NCL14 nebo NCL10. Nástup příznaků nastává ve věku 6‒24 měsíců a dochází k opoždění psychomotorického vývoje, hypotonii, mikrocefalii a křečím. Rychle se rozvíjí postižení zraku vlivem atrofie optiku a makulární degenerace. Většina dětí umírá během první dekády života. Pozdně infantilní forma se manifestuje ve věku 2‒4 let, a to opožděním řeči, křečemi (parciálními i generalizovanými tonicko klonickými absencemi). Následuje rozvoj ataxie, myoklonů, regresu psychomotorického vývoje a ztráta zraku kolem 5.‒6. roku. Nejčastěji nacházíme mutace v genu pro NCL2, vzácněji pro typy NCL5‒8. Juvenilní forma má nástup příznaků mezi 4.‒10. rokem života. Prvním symptomem je obvykle postižení zraku, kdy se slepota rozvine během 2‒3 let. Epilepsie nastupující mezi 5.‒18. rokem může být od parciální přes myoklonickou až po generalizované tonicko klonické křeče. Porucha řeči zahrnuje dysartrii a echolalii, běžně se objevují poruchy chování a rozvoj mentální retardace. Geneticky je podmíněna mutacemi v genech pro NCL3, méně často mutace NCL9 nebo NCL12. Adultní forma obvykle začíná kolem 30. roku života progresivní myoklonickou epilepsií, mentální retardací a poruchou motorických funkcí. Zrak je zachován. Dědičnost je nejčastěji autozomálně recesivní, ale byly popsány vzácné případy s autozomálně dominantním typem dědičnosti. Nejčastěji nacházíme mutace v genu pro NCL4, vzácněji pro typy NCL11 a NCL13 [5,6].
Sfingolipidózy jsou velmi početnou skupinou onemocnění, pro něž je typické střádání sfingolipidů, složených (glyko)lipidů (tab. 1) [5,6].
Gaucherova choroba je jedním z nejčastějších LSD. Na základě klinických projevů rozlišujeme tři podtypy nemoci, ale ve skutečnosti jde o jedno onemocnění s kontinuálním spektrem příznaků. Nejčastější je non neuronopatický typ 1, který se manifestuje v dětském i v dospělém věku a vyznačuje se výraznou splenomegalií, postižením kostí, pancytopenií bez neurologických příznaků. Akutní neuronopatický typ 2 se projevuje již v kojeneckém věku neprospíváním, organomegalií, očním postižením (strabismus, nystagmus) a rychle progredujícím neurologickým postižením. Subakutní neuronopatický typ 3 je pomalejší formou typu 2 s nástupem v pozdějším věku, především v adolescenci.
Fabryho nemoc je X vázané pomalu progredující onemocnění, jež s různou intenzitou postihuje řadu tkání, především cévy, ledviny a myokard. Začíná v dětství a u neléčených jedinců může vést až k renálnímu selhání s nutností hemodialyzační léčby nebo transplantace ledvin. Postižení myokardu se obvykle objevuje v dospělém věku a zahrnuje hypertrofické změny obou komor a následně projevy srdečního selhání.
Krabbeho nemoc je enzymopatie způsobená deficitem enzymu nebo jeho aktivátoru saposinu A, což vede k akumulaci galaktosylsfingosinu (psychosinu) zejména v oligodendrogliích a ve Schwannových buňkách. Klinicky rozlišujeme čtyři formy: časně infantilní (se začátkem do půlroku života, charakterizovaná opožděním vývoje, ztrátou zraku, iritabilitou, hypertonií a decerebrační rigiditou), pozdně infantilní (s nástupem v kojeneckém věku), juvenilní (se začátkem příznaků do věku 10 let s opožděním vývoje až regresem, spastickou tetraparézou, ataxií a atrofií zrakových nervů) a adultní (vzácná, s projevy spastické paraparézy a mozečkové symptomatologie).
Metachromatická leukodystrofie je zapříčiněna dysfunkcí enzymu nebo jeho aktivátoru (saposinu B), čímž dochází k poruše degradace sulfoglykolipidů, které jsou obsaženy v myelinu neuronů centrálního i periferního nervového systému. Rozeznáváme infantilní formu s nástupem zejména svalové slabosti, neobratnosti, dyspraxie, poruch chůze a řeči nebo epilepsie ve věku kolem 1–2 let. Následně se rozvíjí spasticita, porucha zraku při atrofii zrakových nervů a porucha sluchu. Juvenilní forma nastupuje do věku 15 let a manifestuje se poruchou pozornosti, zhoršením školního prospěchu, poruchou chůze. U adultní formy dominují neurologické a psychiatrické symptomy.
U Niemannovy‒Pickovy choroby typu A/B dochází k hromadění sfingomyelinu v buňkách retikuloendoteliálního systému, v hepatocytech a plicních makrofázích. Jako formu A (NPA) označujeme závažnější průběh nemoci, kdy se v kojeneckém věku rozvíjí závažná hepatosplenomegalie a mírná kraniofaciální dysmorfie (antevertované nostrily, širší kořen nosu). Zpočátku je psychomotorický vývoj bez nápadností, ale následně dochází k jeho zpomalení až regresu; progreduje i axiální a kořenová hypotonie s akrální spasticitou. Střádání v retinálních gangliových buňkách sítnice vede k obrazu „třešňové skvrny“. Mírnější forma B (NPB) se manifestuje od batolecího po dospělý věk, nejčastěji hepatosplenomegalií. U dětí se projevuje porucha růstu. Pozvolna progreduje restriktivní plicní postižení, jaterní cirhóza a kardiální selhání. Neurologické a oční postižení není u NPB vyjádřeno v takové míře.
Všechny formy GM1 gangliosidózy jsou podmíněny dysfunkcí enzymu vedoucí k hromadění jak gangliosidů, tak i dalších glykosfingolipidů, oligosacharidů a GAG a proto se u nejtěžších forem kombinují současně příznaky sfingolipidóz, MPS a oligosacharidóz. I u této nemoci rozlišujeme typickou časně infantilní formu (typ I) s nástupem hypotonie během prvních dnů života. Následují problémy s krmením, zástava neurologického vývoje, rozvoj hepato/splenomegalie, ztráta zraku a dysostotické změny na skeletu. Hypotonie se mění na spasticitu a dochází k regresu neurologického vývoje s křečemi. Pozdně infantilní forma (typ II) se rozvíjí v kojeneckém věku a zahrnuje poruchu chůze, regres vývoje, spastickou kvadruparézu a křeče. Pacienti netrpí hepatosplenomegalií ani kraniofaciální dysmorfií. Jako adultní forma jsou označovány chronické varianty s pozdním začátkem, kde dominují neurologické obtíže (dysartrie, dystonie).
GM2 gangliosidózu tvoří tři biochemicky i geneticky odlišné nemoci: Tayova‒Sachsova choroba (defekt α podjednotky, tedy deficit hexosaminidázy A); Sandhoffova choroba (defekt β podjednotky, tedy deficit hexosaminidázy A i B) a deficit GM2 aktivátoru hexosaminidázy. Všechny tyto tři formy vedou k poruše degradace GM2 gangliosidu. Klinicky rozlišujeme infantilní formu, u níž se hypotonie a typická úleková reakce na zvuk s extenzí paží objevuje do 6. měsíce života. Rozvíjí se slepota, spasticita, křeče a poruchy polykání, někdy hepatosplenomegalie. Typickým znakem je nález na sítnici charakteru třešňové skvrny. Pozdně infantilní a juvenilní formy začínají do věku 10 let ataxií, nekoordinovanými pohyby a dysartrií s následným rozvojem psychomotorické retardace, spasticity a křečí. Pro adultní formu jsou typické neurologické projevy (pyramidové i extrapyramidové s dystonií, atetózou či ataxií) a psychotické obtíže.
Mezi další LSD patří Wolmanova choroba, Niemannova‒Pickova choroba typu C, Pompeho a Danonova nemoc nebo cystinóza se střádáním lipidů, glykogenu nebo cystinu.
Wolmanova choroba a její mírná forma označovaná jako CESD (cholesterol ester storage disease) jsou způsobeny enzymovým defektem lysozomální kyselé lipázy. Dochází k hromadění esterů cholesterolu a triglyceridů v lysozomech mnoha tkání. Nemoc se klinicky manifestuje již v novorozeneckém věku průjmy, zvracením, steatoreou, neprospíváním, hepatosplenomegalií nebo psychomotorickou retardací. Typicky nacházíme zvětšení nadledvinek s kalcifikacemi. Pro CESD je typický nástup v dospělém věku s hepatomegalií, jaterním postižením s hepatopatií, lymfadenopatií a dyslipidemií.
Niemannova‒Pickova choroba typu C (NPC) představuje klinicky velmi heterogenní onemocnění, které se může manifestovat od novorozeneckého věku až po pozdní dospělost. Příčinou NPC je funkční porucha lysozomálního membránového proteinu NPC1 (95 % případů) nebo solubilního lysozomálního proteinu NPC2 (méně než 5 % případů), které se oba podílejí na transportu cholesterolu a dalších lipidových látek z lysozomu/pozdního endozomu do endoplazmatického retikula, Golgiho aparátu a na plazmatickou membránu. Porucha vede k hromadění neesterifikovaného cholesterolu, sfingomyelinu a dalších glykosfingolipidů v lysozomech. Klinicky rozeznáváme vzácnou neonatální formu, časnou a pozdní infantilní formu, juvenilní a adultní formu NPC. Infantilní formy jsou méně časté a příznaky se objevují ve věku mezi dvěma měsíci až dvěma roky. Zahrnují neurologické obtíže (poruchu chůze, spasticitu, opožděný vývoj řeči, centrální hypotonii, gelastickou kataplexii aj.) a splenomegalii. Většinou je přítomna vertikální supranukleární pohledová obrna (VSPO). Juvenilní forma často označovaná jako klasická forma je charakterizována nenápadným rozvojem neurologických příznaků ve školním věku, mezi něž patří poruchy učení, dystonie, dysfagie, poruchy pozornosti a aktivity, dyspraxie a poruchy artikulace; dále splenomegalií a popisovány jsou VSPO, kataplexie a narkolepsie. Adultní forma se objevuje od věku 15 let a má podobné příznaky jako juvenilní forma, ale s pozdějším nástupem. Dominují zde neuropsychiatrické obtíže s poruchou kognitivních i exekutivních funkcí. I u adultní formy nacházíme splenomegalii, která však nemusí být tak výrazná.
Pompeho choroba, glykogenóza typu II, je zapříčiněna poruchou lysozomálního enzymu, který hydrolyticky štěpí glykosidické vazby glykogenu, jenž se následně hromadí zejména ve svalech a srdci. Infantilní forma se nejčastěji manifestuje povšechnou hypotonií (tzv. obraz floppy baby), neprospíváním, kardiomegalií a hypertrofickou kardiomyopatií. Juvenilní a adultní typ se projevuje progredující svalovou slabostí, proximální pletencovou myopatií a respirační insuficiencí. Srdeční postižení není natolik dominantním znakem.
Danonova choroba je dalším LSD s poruchou metabolismu glykogenu, u kterého je defektní protein LAMP2 (lysosomal associated membrane protein 2), jehož přesná funkce není dosud známa, nicméně předpokládá se, že hraje důležitou roli v autofagii. Klinicky se prezentuje u mladších mužů těžkou hypertrofickou kardiomyopatií. Často jsou přítomny poruchy srdečního rytmu, různá tíže svalové slabosti a mentální retardace. Oční vyšetření může odhalit retinopatii nebo makulopatii [7].
Cystinóza je způsobena poruchou membránového proteinu cystinosinu, jehož funkcí je transport cystinu z lysozomu, který se zde tvoří při katabolismu proteinů. Nemoc má tři různé klinické fenotypy, přičemž nejčastější je nefropatická neboli infantilní forma, pro kterou je charakteristický nástup obtíží v kojeneckém věku. Typickými příznaky jsou neprospívání, polyurie, Fanconiho syndrom. U neléčených pacientů může renální postižení vyústit do konečné fáze selhání ledvin ve věku mezi 6‒12 lety. Patologickým podkladem je nápadná atrofie ledvin s glomerulární sklerózou a s tubulointersticiální fibrózou. V batolecím věku dochází k rozvoji rachitických změn a dalších extrarenálních komplikací. Mezi oční symptomy patří fotofobie a akumulace depozit cystinových krystalů v rohovce, které mohou u neléčených pacientů vést až ke slepotě. Hromadění cystinu v extrarenálních orgánech ukazuje na multisystémový průběh cystinózy a zahrnuje postižení štítné žlázy, jater, sleziny, pankreatu, svalů a centrální nervové soustavy. Jsou popisovány příznaky hypotyreózy, gonadální dysfunkce zejména u chlapců, inzulin dependentní diabetes mellitus, hepatosplenomegalie, myopatie a velmi výjimečně cystinotická encefalopatie. Juvenilní forma cystinózy je mírnější verze infantilní formy, kde jsou příznaky vyjádřeny v pozdějším věku a s menší intenzitou. Další formou cystinózy je adultní benigní forma, pro niž je typické izolované oční postižení při hromadění cystinových krystalů [8].
Poruchy kobalaminového metabolismu (CblF a CblJ) jsou extrémně vzácná onemocnění, u nichž nastává porucha transportu kobalaminu přes lysozomální membránu. Kobalamin se tak hromadí v lysozomech a nemůže být v cytosolu metabolizován na adenosylkobalamin a metylkobalamin, což jsou kofaktory pro důležité enzymy – metylmalonyl CoA mutázu a metioninsyntázu. Pro pacienty jsou charakteristické hematologické nálezy megaloblastové/makrocytární anémie, neutropenie, trombocytopenie, a dále neprospívání, opoždění psychomotorického vývoje, hypotonie a hyperpigmentace, které se objevují již v dětském věku.
Diagnostika lysozomálních střádavých
onemocnění
Diagnostika LSD je podložena klinickým podezřením, a to nejčastěji u pacientů s progredujícím multisystémovým postižením, jež podporují nálezy ze zobrazovacích vyšetření – organomegalie na ultrasonografickém vyšetření nebo dysostotické změny při rentgenovém vyšetření.
Nespecifické enzymologické markery LSD
Laboratorně můžeme využít některých nespecifických markerů, jako jsou kyselá fosfatáza nebo chitotriosidáza, jejichž enzymová aktivita se u pacientů s LSD zvyšují stimulací makrofágového systému. Zvýšenou aktivitu chitotriosidázy v séru či v plazmě nacházíme u většiny pacientů s Niemannovou‒Pickovou chorobou typu A/B a typu C a u pacientů s Gaucherovou chorobou. Ojediněle může být mírně zvýšená hodnota i u pacientů s Krabbeho leukodystrofií, Wolmanovou chorobou/CESD, GM1 gangliosidózou, cystinózou, alfa mannosidózou, MPS typu IV i IVB. Je nutné si uvědomit, že normální aktivita chitotriosidázy nevylučuje diagnózu LSD. Přibližně 5 % populace má však deficit chitotriosidázy, který je bez klinického korelátu.
Histopatologické nálezy
Při vyšetření nátěru periferní krve můžeme u pacientů s LSD prokázat známky střádání v leukocytech. Vakuolizaci v lymfocytech nacházíme u některých pacientů s LSD, kteří trpí GM1 gangliosidózou, Niemannovou‒Pickovou chorobou typu A/B, fukosidózou, alfa mannosidózou, galaktosialidózou, sialidózou, Wolmanovou chorobou, mukolipidózou typu II, III nebo IV nebo neuronální ceroidlipofuscinózou 3 (tab. 3). U nemocných s různými MPS nacházíme ojediněle neutrofilní granula v leukocytech. U některých pacientů s organomegalií či s hematologickými změnami je prováděna v rámci širší diferenciální diagnostiky i punkce kostní dřeně. Nález střádavých buněk ve vzorku kostní dřeně je typický pro Gaucherovu a Niemannovu‒Pickovou chorobu (NPA/B i NPC), ale můžeme je nalézt rovněž u nemocných s GM1 gangliosidózou, Farberovou chorobou, fukosidózou, alfa mannosidózou nebo s galaktosialidózou (tab. 3). V diagnostice NCL jsou využívány charakteristické histopatologické změny ve vzorcích z kožní biopsie – granulární osmiofilní depozita nacházíme u NCL1 (u vzácné NCL10), kurvilineární profily u NCL2 a NCL7, obraz otisku prstu u NCL3. Kombinace těchto znaků jsou popisovány u NCL5‒8 a u adultních forem NCL4, NCL11 a NCL13. U pacientů s Pompeho chorobou, kteří podstoupí v rámci diferenciálně diagnostického procesu svalovou biopsii, je popisován histochemický průkaz akumulace glykogenu ve svalu a nález vakuolární degenerace svalových vláken. U závažnějších postižení je popisována fibrotizace a atrofie svalových vláken. Naopak u přibližně 25 % pacientů není ve svalu nalezena jednoznačná akumulace glykogenu, jelikož postižení jednotlivých svalů může být nerovnoměrné a nehomogenní. U pacientů s Danonovou chorobou nacházíme ve svalové biopsii obraz vakuolární myopatie s vakuolami obsahujícími glykogen.
Biochemické vyšetření metabolitů
U nemocných s podezřením
na některý z typů MPS je na prvním místě
vyšetření GAG v moči (dříve označovaných jako
mukopolysacharidy; tab. 3).
Analýza GAG v moči je prováděna jak kvantitativně, tak
i kvalitativně. Ve vzorku ze sběru moče nebo
z ranní porce moče se stanovuje celkové množství GAG
a následným elektroforetickým vyšetřením se dále typizují
jednotlivé GAG. Podle profilu zastoupení jednotlivých frakcí GAG
můžeme více specifikovat enzymologické vyšetření. Pro MPS typu
I, II, VI a VII je typické zastoupení heparansulfátu
a dermatansulfátu ve dvou frakcích. U pacientů
s MPS typu III nacházíme izolovaně zvýšené vylučování
heparansulfátu, naopak u MPS typu IV je přítomen
keratansulfát i chondroitinsulfát. U vzácné MPS typu IX
je přítomna hyaluronová kyselina v moči. Vylučování GAG
do moče závisí na věku vyšetřovaného a se
zvyšujícím se věkem dochází ke snížení exkrece, což
u starších pacientů může vést k falešně negativním
výsledkům [2,3].
vyšetřením se dále typizují
jednotlivé GAG. Podle profilu zastoupení jednotlivých frakcí GAG
můžeme více specifikovat enzymologické vyšetření. Pro MPS typu
I, II, VI a VII je typické zastoupení heparansulfátu
a dermatansulfátu ve dvou frakcích. U pacientů
s MPS typu III nacházíme izolovaně zvýšené vylučování
heparansulfátu, naopak u MPS typu IV je přítomen
keratansulfát i chondroitinsulfát. U vzácné MPS typu IX
je přítomna hyaluronová kyselina v moči. Vylučování GAG
do moče závisí na věku vyšetřovaného a se
zvyšujícím se věkem dochází ke snížení exkrece, což
u starších pacientů může vést k falešně negativním
výsledkům [2,3].
Diagnostika glykoproteinóz je založena na detekci oligosacharidů v moči, které slouží jako screeningová metoda (tab. 3). Používá se tenkovrstevná chromatografie s následnou detekcí různými činidly. Nově se rozvíjí analýza pomocí tandemové hmotnostní spektrometrie. Limitující může být snížená exkrece oligosacharidů nebo sialové kyseliny do moče u starších pacientů, kteří by tak mohli uniknout diagnóze [5,6].
U metachromatické leukodystrofie dochází k hromadění sulfatidů v epitelu buněk distálního tubulu ledvin, což vede i k jejich vylučování do moče, kde je lze detekovat pomocí vysokoúčinné kapalinové chromatografie. Na principu reakce sulfatidů s některými pozitivně nabitými barvivy za vzniku barevného posunu, tzv. metachromazie, je založena screeningová metoda detekce tzv. metachromatických substancí v moči. Limitujícím faktorem zůstává nutnost sběru moče a analýza v dobře rozmíchaném vzorku obsahujícím i případné sedimentované částice.
Diagnostickou metodou u pacientů s podezřením na Niemannovu‒Pickovu chorobu byl tzv. filipinový test, který je vysoce specifický a je založený na průkazu intracelulárních depozit neesterifikovaného cholesterolu ve fibroblastech z kožní biopsie. Méně specifický je test rychlosti esterifikace značeného LDL cholesterolu ve fibroblastech. Od těchto testů se postupně upouští pro nutnost invazivního postupu (kožní biopsie), a využívají se proto ve specifických případech (např. prokázání patogenicity u nálezu nových mutací). Výjimečně se stanovuje koncentrace vybraných oxysterolů (zejména cholestantriolu a 7 ketocholesterolu). Nově je dostupná senzitivní a specifická screeningová metoda založená na stanovení metabolitů lysosfingomyelinu 509 (SPC509) a lysosfingomyelinu (SPC) v séru/plazmě či v suché krevní kapce. Hodnota SPC509 je signifikantně zvýšena v plazmě jak u pacientů s NPA/B, tak rovněž s NPC a nelze je podle výše odlišit, zatímco hodnota SPC je zvýšena pouze u NPA/B. V případě analýzy ve vzorku suché krevní kapky se využívá SPC509, jehož hodnota je zvýšená u obou chorob [6,9].
Enzymologická vyšetření
Zlatým standardem v diagnostice LSD je specifické enzymologické vyšetření (tab. 3). V tabulce 1 jsou uvedeny názvy enzymů u každého onemocnění, které je způsobeno defektem enzymu. Enzymologická analýza se nejčastěji provádí v leukocytech periferní krve, nicméně je dostupná i v dalších buňkách, např. kožních fibroblastech. Nově je zaváděna i analýza v plazmě nebo ve vzorcích suché krevní kapky, což výrazně zlepšuje možnosti analýzy i v zaslaných vzorcích, kde je nižší pravděpodobnost rizika inaktivace vlivem teplot či dalších faktorů během transportu. V rámci prenatální diagnostiky se běžně provádí enzymologická analýza v kultivovaných amniocytech nebo v buňkách choriových klků. V některých zemích již probíhají pilotní studie na diagnostiku vybraných LSD v rámci novorozeneckého laboratorního screeningu, kdy se využívá enzymologická analýza ve vzorku suché krevní kapky novorozence.
U některých LSD se setkáváme s tzv. pseudodeficitem, kdy mutace v genu nevede k dostatečnému snížení aktivity enzymu in vivo, a tím nedochází ke klinickým projevům. Naopak je snížena afinita k syntetickému substrátu používanému v rámci enzymologické analýzy in vitro. V případě metachromatické leukodystrofie jsou v evropské populaci až 2 % nosičů pseudodeficitní alely. Dalšími příklady je Krabbeho leukodystrofie nebo Tayova‒Sachsova choroba.
Zvláštní roli hraje enzymová analýza v diagnostice mukolipidóz typu II a III. Jelikož se jedná o poruchu v posttranslační glykosylaci lysozomálních enzymů, nedochází tak k jejich správnému transportu do buněk. Zůstávají tedy v extracelulárním prostoru, čehož se využívá v analýze, kdy detekujeme jejich zvýšenou aktivitu v plazmě (např. hexosaminidázy a alfa mannosidázy).
Molekulárně genetická vyšetření
V rámci potvrzení diagnózy se hojně využívají molekulárně genetická vyšetření. Tabulka 1 obsahuje seznam genů odpovídajících jednotlivým LSD. Vyšetřením familiárních mutací v daném genu se ověřuje prenatální diagnostika anebo se využívá při genetickém poradenství v rodině postiženého u asymptomatických příbuzných. U některých LSD byl popsán tzv. pseudogen, který může mít vysoký podíl homologních sekvencí DNA se skutečným genem, a tak představovat problém při genetickém vyšetření. Pseudogeny jsou známy pro geny GBA (Gaucherova choroba) nebo IDS (MPS typu II).
Vyšetření u dalších LSD
Prvním laboratorním nálezem u kobalaminových defektů je snížené množství vitaminu B12 v krvi a dále nález hyperhomocysteinemie. Speciální metabolická vyšetření ukážou zvýšené vylučování metylmalonátu v moči, což může velmi často vést k chybné diagnóze deficitu vitaminu B12 (při malnutričních příčinách, vegetariánství atd.). Komplementační studie ve fibroblastech a molekulárně genetické vyšetření genů diagnózu kobalaminových defektů potvrdí.
Diagnóza cystinózy, ale i monitorace léčby u pacientů je založena na vyšetření cystinových krystalů hromadících se v leukocytech. Vyšetřením předního segmentu oka pomocí štěrbinové lampy lze odhalit hromadění krystalků cystinu v rohovce.
Závěr
Lysozomální střádavá onemocnění jsou velkou skupinou chorob s různou klinickou prezentací, věkem nástupu i tíží příznaků. Tato přehledová práce předkládá souhrn klinických a laboratorních nálezů u pacientů s jednotlivými LSD. Diagnostické přístupy jsou shrnuty podle analytického přístupu. Právě včasné stanovení diagnózy umožní zahájit jak vhodnou léčbu, tak i případné genetické poradenství v rodině.
Seznam použité literatury
- [1] Poupětová H, Ledvinová J, Berná L, et al. The birth prevalence of lysosomal storage disorders in the Czech Republic: comparison with data in different populations. J Inherit Metab Dis 2010; 33: 387‒396.
- [2] Parenti G, Wraith EJ. The Mucopolysaccharidoses in Physicianʼs Guide to the Diagnosis, Treatment, and Follow‑Up of Inherited Metabolic Diseases. In Blau N, Duran M, Gibson KM (eds.): Laboratory Guide to the Methods in Biochemical Genetics. Berlin, Heidelberg: Springer, 2014; p. 449‒464.
- [3] Jones S, Wijburg F. Mucopolysaccharidoses, Oligosaccharidoses and Sialic Acid Disorders. In Saudubray JM, Baumgartner JM, Walter J (eds.): Inborn Metabolic Diseases. Diagnosis and Treatment. Berlin, Heidelberg: Springer, 2016; p. 121‒137.
- [4] Lukacs Z, Beck M. Oligosaccharidoses and Sialic Acid Disorders in Physicianʼs Guide to the Diagnosis, Treatment, and Follow‑Up of Inherited Metabolic Diseases. In Blau N, Duran M, Gibson KM (eds.): Laboratory Guide to the Methods in Biochemical Genetics. Berlin, Heidelberg: Springer, 2014; p. 437‒448.
- [5] Hollak C, Kettwig M, Schlotawa L, et al. Lysosomal Storage Disorders Including Neuronal Ceroid Lipofuscinoses in Physicianʼs Guide to the Diagnosis, Treatment, and Follow‑Up of Inherited Metabolic Diseases. In Blau N, Duran M, Gibson KM (eds.): Laboratory Guide to the Methods in Biochemical Genetics. Berlin, Heidelberg: Springer, 2014; p. 399‒435.
- [6] Vanier MT, Cailland C, Levade T. Disorders of Sphingolipid Synthesis, Sphingolipidoses, Niemann‑Pick Disease Type C and Neuronal Cerois Lipofusciones in Inborn Metabolic Diseases Diagnosis and Treatment. In Saudubray JM, Baumgartner JM, Walter J (eds.): Inborn Metabolic Diseases. Diagnosis and Treatment. Berlin, Heidelberg: Springer, 2016; p. 551‒575.
- [7] Walter J, Labrune P, Laforet P. The Glycogen Storage Diseases and Related Disorders in Inborn Metabolic Diseases Diagnosis and Treatment. In Saudubray JM, Baumgartner JM, Walter J (eds.): Inborn Metabolic Diseases. Diagnosis and Treatment. Berlin, Heidelberg: Springer, 2016; p. 577‒590.
- [8] Niaudet P. Cystinosis in Inborn Metabolic Diseases Diagnosis and Treatment. In Saudubray JM, Baumgartner JM, Walter J (eds.): Inborn Metabolic Diseases. Diagnosis and Treatment. Berlin, Heidelberg: Springer, 2016; p. 623‒629.
- [9] Kuchar L, Sikora J, Gulinello ME, et al. Quantitation of plasmatic lysosphingomyelin and lysosphingomyelin‑509 for differential screening of Niemann‑Pick A/B and C diseases. Anal Biochem 2017; 525: 73‒77.