Primární vaskulitidy – vzácná a opomíjená onemocnění
Souhrn:
Termín „vaskulitida“ označuje zánětlivé onemocnění cév vedoucí k destrukci cévní stěny, následné proliferaci a k jejich uzávěru. Podmínkou pro stanovení diagnózy je, aby cévní stěna byla primárním místem patologického procesu. Jako důsledek vznikají klinické syndromy na podkladě ischemie tkání zásobovaných poškozenými cévami a na podkladě celkových projevů, které provázejí zánětlivé onemocnění. Vaskulitida může vzniknout de novo jako primární postižení cévní stěny neznámé etiologie, častěji se objeví sekundárně po podání léků, při některých infekčních onemocněních, nádorech a difuzních onemocněních pojiva. Tyto manifestace se obvykle liší svým původem, rozsahem a kalibrem postižených cév. Tento přehledný článek je věnován nejčastějším primárním vaskulitidám, jejich diagnostice a léčbě.
Key words: vasculitis – arteriitis – angiography – biopsy – immunosuppression.
Summary:
By term “vasculitis” inflammatory disease of blood vessels is designated that leads to vessel wall destruction followed by proliferation and occlusion of their lumina. Basic condition for this diagnosis is that vessel wall is a primary site of the pathological process. Clinical syndromes are a consequence of this process resulting into ischaemia of tissues supplied by the affected vessels and with constitutional symptoms associated to the inflammatory disease. Vasculitis can occur de novo as a primary involvement of vessel wall of unknown etiology or it develops secondary to drug administration, infectious diseases, tumours and different connective tissue diseases. These manifestations usually vary in their origin, extent and diameter of involved vessels. This review describes the most common primary vasculitides, their diagnostics and management.
Úvod
Termín „vaskulitida“ označuje zánětlivé onemocnění cév vedoucí k destrukci cévní stěny, následné proliferaci a k uzávěru cév. Podmínkou pro stanovení diagnózy je, aby cévní stěna byla primárním místem patologického procesu [1]. Jako důsledek vznikají klinické syndromy na podkladě ischemie tkání zásobovaných poškozenými cévami a celkových projevů, které provázejí zánětlivé onemocnění (horečka, úbytek hmotnosti, anorexie).
Vaskulitida může vzniknout
de novo jako primární postižení cévní stěny neznámé
etiologie, nebo se objeví sekundárně při některých infekčních
onemocněních, nádorech a jiných procesech. Také některá
difuzní onemocnění pojiva jako revmatoidní artritida, systémový
lupus erythematodes, systémová sklerodermie, dětská
dermatomyozitida mohou být provázena vaskulitidou. Tyto manifestace
se liší svým původem, rozsahem a kalibrem postižených cév
[2].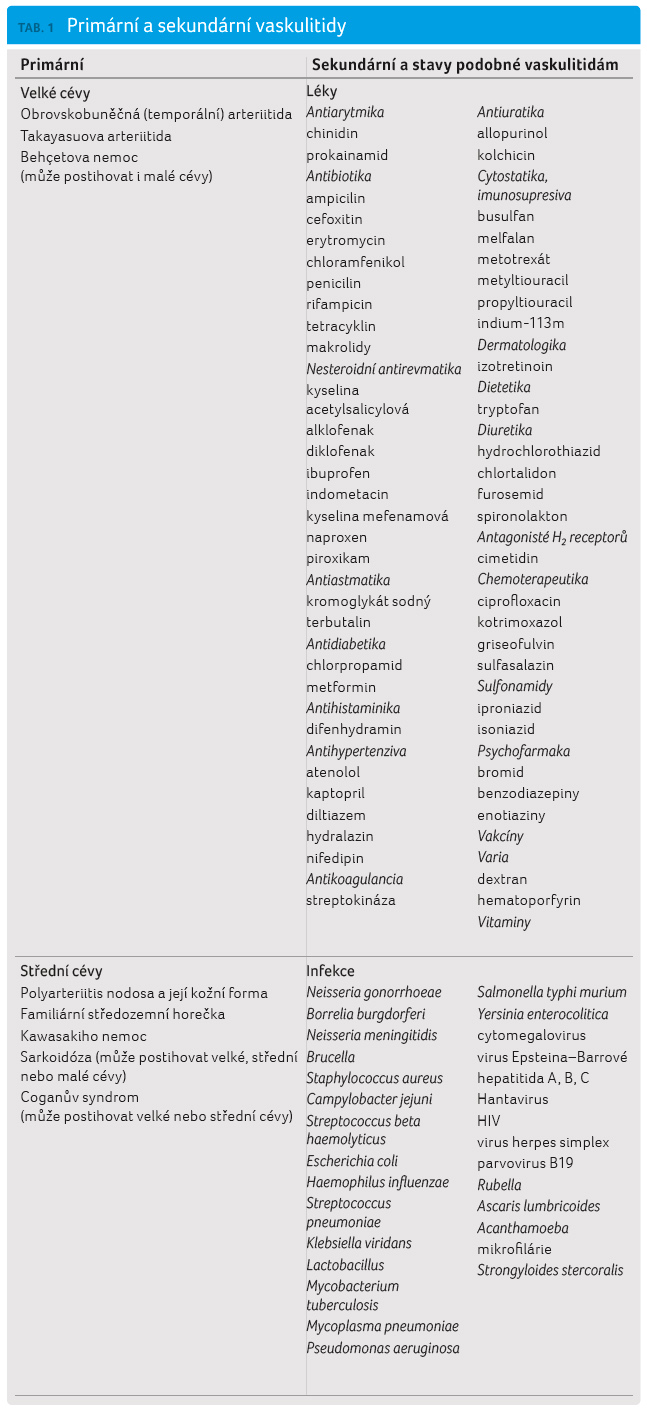
Klasifikace vaskulitid představuje složitý problém, neboť příčina je většinou neznámá a klinické obrazy jsou velmi variabilní a mohou se překrývat. Konečná diagnóza je zpravidla dána histologickým nálezem z biopsie, v některých případech stačí RTG vyšetření s kontrastem nebo jiná zobrazovací metoda. V posledních desetiletích vzniklo několik klasifikací vytvořených v American College of Rheumatology (ACR). Jedna z posledních všeobecně používaných klasifikací pochází z konference v Chapel Hill z roku 1993. Hlavním kritériem bývá kalibr postižených cév. Nejnovější klasifikace Balla upravená Fesslerem [3] je kombinací klasifikace ACR a závěrů konference v Chapel Hill a zahrnuje primární a sekundární vaskulitidu a stavy připomínající vaskulitidy (tab. 1). Tento přehledný článek je věnován nejčastějším primárním vaskulitidám, jejich diagnostice a léčbě.
Obrovskobuněčná arteriitida
Obrovskobuněčná arteriitida (OBA), nazývaná též Hortonova, temporální, je vaskulitida neznámého původu postihující větve zevní i vnitřní karotidy u osob středního a vyššího věku. Toto onemocnění postihuje téměř výhradně jedince bílé rasy starší 50 let s převahou žen. Existují značné rozdíly v incidenci podle geografických oblastí. Nejvyšší počty byly zjištěny na severu USA a v severní Evropě. U poloviny neléčených dochází k oslepnutí bez rozdílu rasy.
Příčina vzniku OBA je dosud nejasná. Výskyt omezený na osoby starší 50 let by ukazoval na souvislost s procesem stárnutí, ale mechanismus vzniku je předmětem hypotéz. Stále se uvažuje o genetické predispozici k této nemoci, neboť u těchto nemocných byla zjištěna zvýšená incidence antigenu HLA DR4 a zejména alel DRB1. Uvažuje se také o roli infekce, protože u části nemocných byly nalezeny zvýšené hodnoty protilátek proti viru parainfluenzy. Spouštěcím agens by mohly být infekce Mycoplasma pneumoniae, Chlamydia pneumoniae nebo infekce parvovirem B19 [4]. Imunologické procesy se nepochybně účastní patogeneze OBA. Zásadní význam mají změny buněčné imunity, zejména cirkulující aktivované monocyty, které secernují prozánětlivé cytokiny. Ve stěně arteria temporalis byly nalezeny CD4+ T lymfocyty a aktivované makrofágy, dále depozita imunoglobulinů a složek komplementu, jež mohou představovat buď protilátky proti stěně tepny, nebo cirkulující imunitní komplexy.
Klinický obraz
Začátek onemocnění je většinou
postupný s rozvojem celkových příznaků (slabost, horečka,
hubnutí), může mít však i akutní průběh. Někdy jsou
tyto příznaky přičítány depresi, stárnutí nebo malignitě. Je
třeba zdůraznit, že příznaky v průběhu doby značně
kolísají i bez léčby. To se týká především bolestí
hlavy, čelistních klaudikací a horečky. Nicméně ztráta
zraku a jiné důsledky uzávěru cév jsou trvalé.
Nejběžnějším příznakem OBA je někdy až úporná cefalea.
Lokalizuje se do jednoho nebo více míst, může být
intermitentní, ale častěji se objevuje denně. Polymyalgia
rheumatica může být jedním z počátečních příznaků,
nebo se objeví později třeba po snížení dávky kortikoidů.
Čelistní klaudikace je typickým příznakem, může postihovat
musculus masseter
nebo temporální svaly, bývá často oboustranná. Objevuje se při
žvýkání tuhých soust. Tyto klaudikace mohou postihnout jazyk
a polykací svaly. Někdy může dojít ke spasmu žvýkacího
svalstva, což zabrání nemocnému v příjmu potravy. Oslabení
pulzu a palpační citlivost spánkových tepen se vyskytuje asi
u poloviny postižených. Je třeba rozlišit, zda se jedná
o citlivost pokožky hlavy a větví arteria
temporali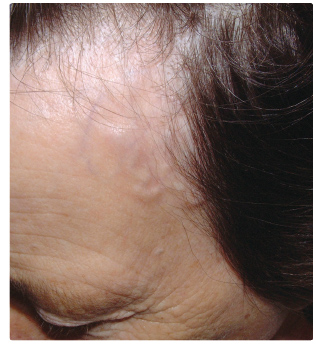 s, nebo svalů a jejich úponů. Zduřelé,
uzlovité nebo jen ztluštělé větve spánkové tepny (obr. 1) jsou jasným příznakem arteriitidy. Segmentální
oslabení pulzu spánkové tepny svědčí pro vaskulitidu, kdežto
celkové oslabení pulzu proximální části karotidy bývá
důsledkem aterosklerotického zúžení. Oční příznaky jsou
časté – amaurosis fugax, jiskřící skotomy a diplopie.
Přechodná porucha vidění bez adekvátní terapie často přechází
do trvalé slepoty. Asi 10 % nemocných má příznaky
postižení velkých tepen končetin manifestující se oslabením
pulzu a klaudikacemi zejména horních končetin, nezřídka
i Raynaudovým fenoménem. Protože se uzávěry velkých tepen
vyvíjejí pomalu, kolaterální oběh může včas zabránit těžké
ischemii. Artritida se popisuje asi u 15 % nemocných.
Synovitida bývá lehká a zpravidla mizí po zahájení
léčby kortikoidy. Postihuje hlavně kolena a kotníky, méně
často zápěstí. Neurologické příznaky jsou pestré –
nejčastěji mononeuropatie nebo periferní polyneuropatie. Vzácné
není ani mozkové krvácení v povodí arteria
carotis interna nebo arteria
basilaris. Dále se objevují tinnitus, vertigo
a jednostranné poruchy sluchu.
s, nebo svalů a jejich úponů. Zduřelé,
uzlovité nebo jen ztluštělé větve spánkové tepny (obr. 1) jsou jasným příznakem arteriitidy. Segmentální
oslabení pulzu spánkové tepny svědčí pro vaskulitidu, kdežto
celkové oslabení pulzu proximální části karotidy bývá
důsledkem aterosklerotického zúžení. Oční příznaky jsou
časté – amaurosis fugax, jiskřící skotomy a diplopie.
Přechodná porucha vidění bez adekvátní terapie často přechází
do trvalé slepoty. Asi 10 % nemocných má příznaky
postižení velkých tepen končetin manifestující se oslabením
pulzu a klaudikacemi zejména horních končetin, nezřídka
i Raynaudovým fenoménem. Protože se uzávěry velkých tepen
vyvíjejí pomalu, kolaterální oběh může včas zabránit těžké
ischemii. Artritida se popisuje asi u 15 % nemocných.
Synovitida bývá lehká a zpravidla mizí po zahájení
léčby kortikoidy. Postihuje hlavně kolena a kotníky, méně
často zápěstí. Neurologické příznaky jsou pestré –
nejčastěji mononeuropatie nebo periferní polyneuropatie. Vzácné
není ani mozkové krvácení v povodí arteria
carotis interna nebo arteria
basilaris. Dále se objevují tinnitus, vertigo
a jednostranné poruchy sluchu.
Stanovení diagnózy
Diagnózu OBA potvrdí histologický nález z biopsie větve arteria temporalis, přičemž je třeba vyšetřit postižený úsek. Pokud není tepna jasně makroskopicky postižena, je vhodné odebrat delší úsek (4–6 cm). Pokud se v prvním vzorku neprokáží patologické změny a trvá klinické podezření, je vhodné provést biopsii na druhé straně. Z neinvazivních vyšetření se užívá Dopplerovo měření průtoku, které může určit místo vhodné pro biopsii při nevýrazném klinickém nálezu. Další metodou je oční pneumopletysmografie, jejíž pomocí lze ověřit snížení průtoku arteria centralis retinae, které může včas odhalit riziko poruchy zraku. Pro stanovení diagnózy OBA byla ACR v roce 1990 vypracována klasifikační kritéria (tab. 2) [5]. Při přítomnosti tří kritérií z pěti je senzitivita 93,5 % a specificita 91,2 %. Většina nemocných OBA má mírnou hypochromní nebo normochromní anémii. Sérová koncentrace ž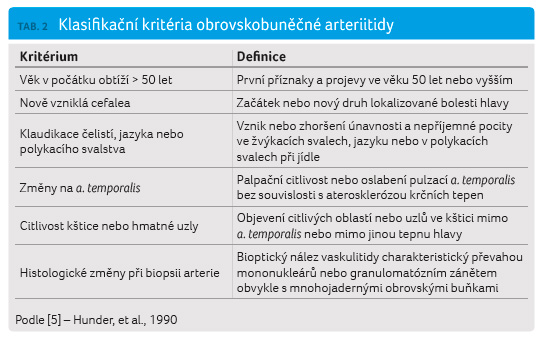 eleza a saturace transferinu bývají
snížené. Počet leukocytů je obvykle v normě, počet
destiček spíše vyšší. Typické jsou vysoké hodnoty reaktantů
akutní fáze zánětu a asi ve čtvrtině případů
nacházíme zvýšení hodnot jaterních testů, kdy je biopticky
zjištěna steatóza až lehká fibróza. Orgánově nespecifické
autoprotilátky bývají negativní. Histopatologický nález ve svém
klasickém obraze ukazuje infiltraci lymfocyty s fragmentací
lamina elastica interna,
dále granulomatózní zánět s histiocyty a mnohojadernými
obrovskými buňkami. Nacházíme T lymfocyty typu CD4+ a příměs
B lymfocytů. Zmnožené retikulocyty jsou důkazem imunitní reakce
probíhající v cévní stěně.
eleza a saturace transferinu bývají
snížené. Počet leukocytů je obvykle v normě, počet
destiček spíše vyšší. Typické jsou vysoké hodnoty reaktantů
akutní fáze zánětu a asi ve čtvrtině případů
nacházíme zvýšení hodnot jaterních testů, kdy je biopticky
zjištěna steatóza až lehká fibróza. Orgánově nespecifické
autoprotilátky bývají negativní. Histopatologický nález ve svém
klasickém obraze ukazuje infiltraci lymfocyty s fragmentací
lamina elastica interna,
dále granulomatózní zánět s histiocyty a mnohojadernými
obrovskými buňkami. Nacházíme T lymfocyty typu CD4+ a příměs
B lymfocytů. Zmnožené retikulocyty jsou důkazem imunitní reakce
probíhající v cévní stěně.
V diferenciální diagnóze obvykle není obtížné odlišit OBA od jiných typů vaskulitid – onemocnění se vyznačuje charakteristickým klinickým obrazem, typickou histologií a orgánovým postižením. Jiné nekrotizující vaskulitidy jen zřídka postihují větve arteria carotis externa. Podobný morfologický obraz je možné pozorovat u Takayasuovy arteriitidy, avšak ta mívá jinou distribuci změn a je v Evropě mnohem vzácnější.
Léčba
Lékem volby OBA jsou kortikosteroidy a léčbu je třeba zahájit co nejdříve, abychom předešli rozvoji cévní mozkové příhody nebo ztrátě zraku. Nikdy nečekáme na nález z biopsie arteria temporalis. Většina autorů doporučuje iniciální dávku prednisonu 40–60 mg denně nebo jiný ekvivalent. Při podezření na výše uvedené manifestace zahajujeme léčbu buď denní dávkou prednisonu 80–120 mg, nebo infuzí metylprednisolonu 500–1 000 mg/den podávanou po tři dny po sobě a poté následuje terapie prednisonem v dávce 60 mg. Většina příznaků vymizí během 1–3 dnů. Po poklesu hodnot ukazatelů zánětu a ústupu reverzibilních příznaků, tj. asi po 2–4 týdnech, je možné dávku prednisonu snížit. Dávka 5–10 mg může většinou udržet arteriitidu pod kontrolou, ale průměrná délka podávání bývá 2–3 roky, avšak u některých nemocných i déle. Ke snížení dávek kortikoidů byla navrhována jako alternativní terapie imunosupresiva – metotrexát, cyklofosfamid a azathioprin, nejsou však k dispozici kontrolovaná klinická hodnocení s těmito léčivy.
V současnosti je snaha podávat při onemocnění OBA biologickou léčbu. Blokátory tumor nekrotizujícího faktoru alfa (TNFα) je možné použít u pacientů s perzistující aktivitou nemoci se dvěma a více vzplanutími při adekvátní léčbě kortikosteroidy a s alespoň jedním inunosupresivem. Podání imunosupresiva není podmínkou, pokud je kontraindikováno nebo nesnášeno. Flare (vzplanutí) je definováno jako klinická manifestace s vysokou hodnotou sedimentace erytrocytů (Fahræus-Westergren, FW) a C reaktivního proteinu (CRP) při léčbě dávkou prednisonu vyšší než 5 mg. Tocilizumab je možné použít u pacientů s postižením velkých cév a s perzistující aktivitou nemoci se dvěma a více vzplanutími při adekvátní léčbě kortikosteroidy a alespoň jedním imunosupresivem. Podání imunosupresiva není podmínkou, pokud je kontraindikováno nebo nesnášeno [6].
Prognóza OBA z hlediska přežívání je všeobecně dobrá. V případě neléčení nemoci však může dojít až u poloviny nemocných ke ztrátě zraku nebo k cévní mozkové příhodě až smrti.
Granulomatóza s polyangiitidou
Granulomatóza s polyangiitidou (GPA) neboli Wegenerova granulomatóza je chronická nekrotizující vaskulitida neznámého původu charakterizovaná granulomatózními lézemi. Plně rozvinutý obraz může být život ohrožující s typickým pulmorenálním syndromem [7].
Prevalence onemocnění se odhaduje na 3–6 pacientů na 100 000 obyvatel a roční incidence činí 7 případů na 100 000 obyvatel. Onemocnění postihuje častěji muže, obvykle mezi 40. a 60. rokem života. Etiologie je neznámá. Byla zjištěna významná imunogenetická asociace s alelami HLA DRB1*04 a DRB*13. V patogenezi se uplatňuje geneticky podmíněná nebo získaná nerovnováha proteáza/antiproteáza, kdy u GPA se jedná o proteinázu 3 (PR3). Proti tomuto enzymu se tvoří ANCA protilátky s imunofluorescencí cytoplazmatického typu (cANCA). Z faktorů zevního prostředí může hrát určitou roli hypersenzitivita na blíže neurčený alergen zejména inhalované substance. Z bakteriálních agens se jedná o Staphylococcus aureus a z virových o parvovirus B19 [8]. Křemík a silikon jsou zřejmě asociovány s rychle progredující glomerulonefritidou při GPA. Dále se uplatňuje abúzus drog (šňupání kokainu vyvolává zánětlivé destrukce), podobně jako tyreostatika (propylthiouracyl, metimazol a karbimazol). Nápadná je souvislost s různými typy malignit.
Klinický obraz
Z klinických projevů jsou časté celkové příznaky – únava, slabost, hubnutí a febrilie. Postižení horních dýchacích cest se projevuje ucpaným nosem, epistaxí, chronickou rýmou a sinusitidou. Granulomatózní zánět může mít destruktivní charakter a vést až k sedlovité deformitě nosu. Může se objevit ulcerózní stomatitida a hyperplastická gingivitida. Časté jsou i záněty zevního zvukovodu – chondritida středouší a převodní porucha sluchu. Při postižení trachey může vzniknout subglotická stenóza. Postižení dolních dýchacích cest signalizuje kašel, hemoptýza či bolest na hrudi. Granulomy se mohou provalit do pleurální dutiny a způsobit ventilový pneumotorax. Postižení ledvin je časté pod obrazem rychle progredující glomerulonefritidy a může vést až k renální insuficienci. Oční symptomatologie je pestrá – konjunktivitida, episkleritida, korneální vředy, retinální vaskulitida, neuropatie nervus opticus až protruze bulbu. Postižení periferních nervů se projevuje nejčastěji jako mononeuritis multiplex. Méně časté je postižení gastrointestinálního traktu, které se projevuje průjmy, krvácením i bolestmi břicha, někdy perforací střeva. Vídáme také artralgie a myalgie, ale i erozivní artritidy jsou možné.
Stanovení diagnózy
Při laboratorním vyšetření zjišťujeme normochromní normocytární anémii, leukocytózu s lehkou eozinofilií, často i trombocytemie. Zpravidla je zrychlená FW, vyšší hodnota CRP a mírná hypergamaglobulinemie, zejména zvýšení hodnot IgA. V časné fázi je obvyklá mikroskopická glomerulární erytrocyturie s malou či střední proteinurií. Renální funkce mohou být snížené. Někdy se prokáže revmatoidní faktor a zvýšená koncentrace cirkulujících imunitních komplexů. Vždy vyšetřujeme cANCA (PR3). Vždy se provádí imunofluorescenční vyšetření a při pozitivitě doplňujeme test komerčním ELISA kitem proti PR3 a případně proti myeloperoxidáze. Doporučuje se provádět opakovaná vyšetření, neboť vzestup titrů ANCA protilátek může signalizovat blížící se relaps [9]. Granulomy v horních dýchacích cestách, vedlejších dutinách a v mozku lze dobře znázornit pomocí magnetické rezonance (MR). V histologickém obraze z biopsie lze prokázat drobné granulomy nebo nekrózy se sekundární infekcí. Na skiagramu snímku hrudníku bývá charakteristické motýlovité zastření, pro přesnější obraz je třeba doplnit vyšetření výpočetní tomografií s vysokým rozlišením. Vedle prchavých infiltrátů lze někdy v alveolech také pozorovat rozsáhlé granulomy s centrálním rozpadem. Angiografické vyšetření může být přínosné při postižení tepen středního kalibru při GPA, protože pomůže odlišit vaskulitické a aterosklerotické léze při ischemii střev nebo hypoperfuzi končetin. Někteří autoři doporučují i bronchoskopii s bronchoalveolární laváží a cytologické vyšetření získané tekutiny. Při renální biopsii se popisuje fokální až difuzní segmentální nekrotizující glomerulonefritida. Histologicky lze někdy zachytit i vaskulitidu drobných cév. V duodenu lze endoskopicky prokázat hemoragie až ulcerace.
Diagnóza se opírá o typický klinický obraz celkových příznaků, postižení horních a dolních dýchacích cest s granulomy, postižení očí a v pozdějších fázích i ledvin. Svůj význam má i průkaz ANCA protilátek, jejich nepřítomnost často svědčí pro limitovanou formu onemocnění. Vhodné je potvrzení diagnózy biopsií z horních cest dýchacích. V diferenciální diagnóze je třeba odlišit jiná pulmorenální onemocnění – zejména Goodpastureův syndrom na základě imunofluorescenčního vyšetření vzorků z renální biopsie. U pacientů s tímto syndromem nacházíme lineární depozita imunoglobulinů na bazální membráně glomerulů. Pro eozinofilní granulomatózu s polyangiitidou je příznačné asthma bronchiale, krevní a tkáňová eozinofilie. U polyarteriitis nodosa je postižení plic mnohem vzácnější než u GPA.
Léčba
Terapie má dva hlavní cíle: agresivně vedenou kombinovanou imunosupresí dosáhnout remise onemocnění a udržovací léčbou předejít relapsu. Pro navození remise se užívá prednison v zahajovací dávce 1 mg/kg nebo ekvivalent společně s cyklofosfamidem nebo metotrexátem. Při stavech ohrožujících život nebo funkci orgánů, jako je krvácení do alveolů nebo rychle progredující glomerulonefritida, se na začátku podává 1 g metylprednisolonu v infuzi po tři dny za sebou. Dávka prednisonu se snižuje o 5–10 mg za den podle aktivity a poté pomaleji s cílem dosáhnout dávky 7,5 mg/den. Někdy je nutné ponechat dávku 5 mg i po celou udržovací fázi. Při generalizované GPA se k navození remise podávají intravenózní pulzy cyklofosfamidu 15–20 mg/kg po třech týdnech obvykle šestkrát. U časné systémové formy se doporučuje metotrexát 0,3 mg/kg/týden [10]. Lokalizované granulomatózní formy lze léčit kombinovaným přípravkem trimetoprim/sulfametoxazol. U refrakterních forem se přidávají vysoké dávky intravenózních imunoglobulinů nebo opakovaná plazmaferéza. V udržovací fázi se podává azathioprin 2 mg/kg/den nebo metotrexát, leflunomid 30–40 mg/den nebo trimetoprim/sulfametoxazol. Z biologických léků se zatím jako nejslibnější jeví rituximab v kombinaci s cyklofosfamidem a kortikosteroidy.
Prognóza závisí na míře postižení ledvin, na výskytu oportunní infekce, z akutních příčin úmrtí to bývá masivní plicní krvácení. Relapsy onemocnění se objevují až u 50 % nemocných i několik let od diagnózy, často ve vazbě na infekci či po snížení dávek terapie. Protilátky ANCA jsou užitečným ukazatelem, který může upozornit na hrozící relaps.
IgA vaskulitida
Henochova–Schönleinova purpura, v odborných kruzích označovaná též názvem IgA vaskulitida (IGV), je vaskulitida drobných tepen, která se vyskytuje v dětském věku a obvykle navazuje na prodělané infekční onemocnění.
Nemoc začíná často u dětí ve věku do deseti let. Roční incidence podle literárních údajů činí 100–200 nemocných na milion obyvatel. Původ nemoci je neznámý. U nemocných IGV byl zjištěn zvýšený výskyt alel HLA DRB1*01 a HLA DRB1*11 [11]. Úloha interkurentní infekce je nepochybná, žádné infekční agens však nebylo prokázáno jako etiologické. Zejména se zvažuje vliv respiračních infekcí. Obecně infekce často předchází rozvoji nemoci a byla nejčastěji prokázána tato agens: streptokoky, Staphylococcus aureus, Escherichia coli, Mycobacterium tuberculosis, Yersinia species, Legionella species, Mycoplasma pneumoniae, virus Epsteina–Barrové, virus hepatitidy B, varicella, adenoviry, cytomegaloviry a parvovirus B19. Nemoc také byla vyvolána očkováním proti tyfu, paratyfu, choleře, žluté horečce a spalničkám. Obdobnou provokující roli může hrát léková či potravinová alergie.
Klinický obraz
Artralgie až oligoartritidy často předcházejí kožním projevům a postihují nožní klouby, kotníky nebo kolena. Kožní exantém začíná jako urtikariální eflorescence na extenzorových stranách končetin, okolo kotníků, loktů a na hýždích, méně často na trupu a obličeji. Během dvou dnů dochází k vývoji typické prominující purpury, která může splývat v ekchymózy. Abdominální symptomatologie mívá charakter intenzivní kolikovité bolesti, někdy nauzey a zvracení, vzácně akutní intususcepce. Gastrointestinální krvácení bývá jen okultní, výjimečně masivní. Bolesti a otok skrota a chámovodu mohou vést k diagnóze testikulární torze. Postižení ledvin se manifestuje proteinurií s hematurií, někdy hypertenzí a oligurií s poklesem renálních funkcí až ledvinnou nedostatečností. Neurologické symptomy zahrnují bolesti hlavy, vzácněji poruchy chování a křeče.
Stanovení diagnózy
V laboratorním obraze zaznamenáváme zvýšení hodnot reaktantů akutní fáze, normochromní normocytární anémii, mírnou leukocytózu a trombocytózu. Hematurie s nálezem válců v sedimentu a s proteinurií je indikací k biopsii ledvin. U některých nemocných byly prokázány vysoké titry revmatoidního faktoru ve třídě imunoglobulinů IgA. Hodnoty C3, C4 a CH50 bývají snížené. Z kožní biopsie nacházíme histologicky leukocytoklastickou vaskulitidu s perivaskulární akumulací polymorfonukleárů v okolí kapilár a venul koria. Imunofluorescenčně lze prokázat IgA, C3 a fibrin. Renální biopsie mívá typický obraz fokálně segmentální proliferativní glomerulonefritidy s proliferací mesangiálních buněk. U starších lézí může být přítomen i různý stupeň glomerulosklerózy. Při hemateméze se provádí gastroskopie, kdy nacházíme na sliznici petechie, eroze i ulcerace. Při kolikách je indikována ultrasonografie a CT břicha [12].
Diagnóza se stanoví na základě typického exantému s histologickým nálezem leukocytoklastické vaskulitidy, bolestí břicha a postižení ledvin. V diferenciální diagnostice je zásadní otázkou, zda se jedná o samotný kožní projev, nebo o součást celkového onemocnění. Je třeba odlišit GPA od mikroskopické polyangiitidy, kde bývají pozitivní ANCA protilátky. Systémový lupus erythematodes odliší pozitivita antinukleárních protilátek a autoprotilátek pro dvouvláknové deoxyribonukleové kyseliny. Purpura je jinak průvodním příznakem mnoha infekčních chorob a polékových reakcí, nebývá však hmatná jako u Henochovy–Schönleinovy purpury.
Léčba
V léčbě na počátku podáváme antibiotika, předcházela li infekce. Bolesti a otoky kloubů tlumíme nesteroidními antirevmatiky. Renální insuficienci, bolesti břicha a krvácení z GIT léčíme symptomaticky. Celkově jsou lékem volby kortikosteroidy, které zřejmě snižují riziko intususcepce a působí preventivně při vzniku nefritidy [13]. Při multiorgánovém postižení je indikována kombinovaná imunosupresivní léčba s cyklofosfamidem nebo azathioprinem, případně cyklosporin A s pulzy metylprednisolonu či plazmaferéza. Prognóza je příznivá a je dána závažností renálního postižení, kdy pouze u 3 % nemocných nastává progrese do terminální renální insuficience.
Kdy myslet na vaskulitidu a jak
postupovat při podezření na toto 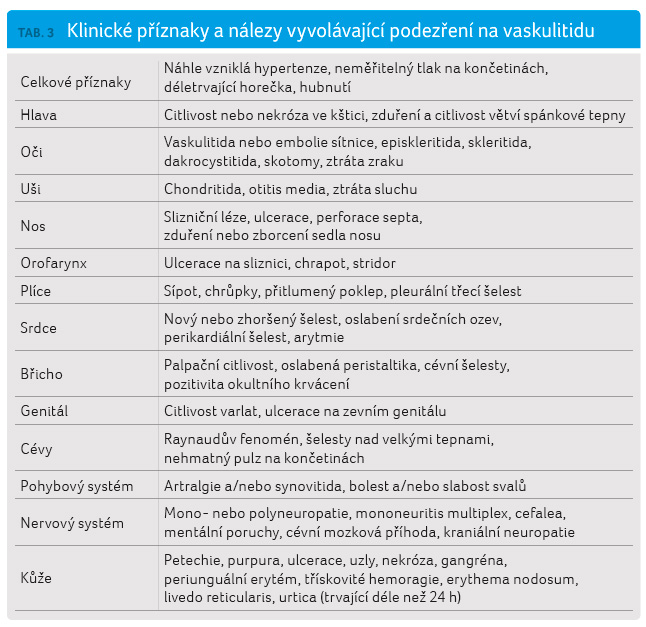 onemocnění
onemocnění
Pro lékaře bývá rozhodující uvědomit si, že některé příznaky a známky, pro něž nemá vysvětlení, mohou být vyvolány probíhajícím zánětlivým procesem v cévní stěně. Klinické projevy v jednotlivých orgánech, které vzbuzují podezření na diagnózu vaskulitidy, jsou uvedeny v tabulce 3 a zahrnují celkové příznaky, jako jsou chronické horečky, projevy ischemie tkání (zejména při neobvyklém věkovém výskytu, jako u mladých žen), neuropatie a podezřelé kožní léze (třeba prominující purpura). Neexistuje jednoduchá prezentace vaskulitidy, proto je zásadní rozpoznání určitých obrazů nebo souborů příznaků. Jak vyplývá z tabulky 3, může být vaskulitidou postižen téměř kterýkoliv orgánový systém. I když se objeví pacient pouze s vyrážkou, musí u něj být vyšetřeny všechny vitálně důležité orgány. Mnohé z těchto příznaků a projevů, pokud se vyskytnou samostatně, jsou nespecifické, ale když jsou interpretovány v kontextu s anamnézou laboratorními nálezy, mohou pomoci stanovit diagnózu. Proto je důležitá znalost základní symptomatologie těchto nemocí.
U vzácných chorob, jako jsou
vaskulitidy, jsou anamnéza a fyzikální vyšetření
nejdůležitějšími diagnostickými nástroji. Základní
algoritmus přístupu k nemocnému při podezření
na vaskulitidu je uveden v obrázku 2. Věk při prvních příznacích může pomoci (OBA
oproti Kawasakiho onemocnění), právě tak mohou hrát roli některé
faktory jako kouření (thrombangiitis obliterans), podané krevní
transfuze, ateroskleróza a výskyt malignit. Předchozí
medikace, cestování, profese, koníčky, potraty, operace
a trombózy představují také důležité informace.
Z laboratorních vyšetření jsou kromě základních testů
vždy důležité výsledky testů na hepatitidy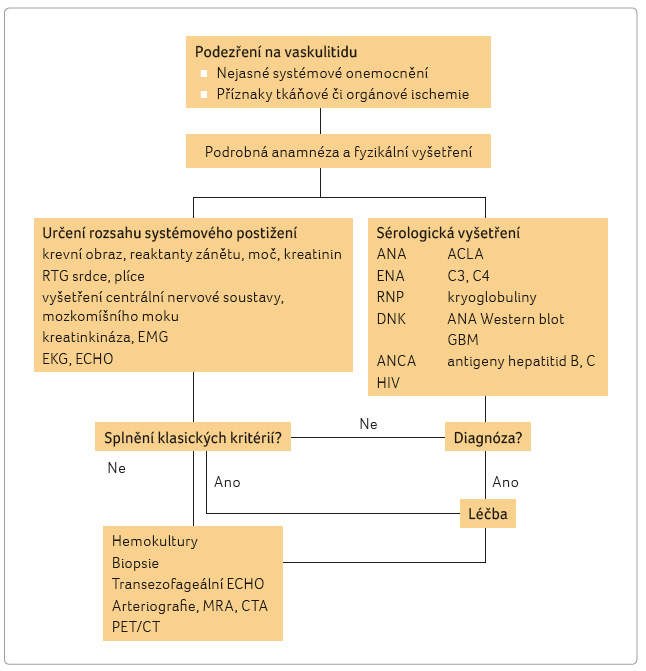 B a C,
HIV a na hodnoty kryoglobulinů. Zásadní význam má
vyšetření ANCA a protilátky proti bazálním membránám
glomerulů, jejichž pozitivita často vede k diagnóze ANCA
asociovaných vaskulitid. Vyšetření antinukleárních protilátek,
autoprotilátek proti deoxyribonukleové kyselině,
ribonukleoproteinu, kardiolipinu slouží k diferenciální
diagnóze jiných běžnějších difuzních onemocnění pojiva,
případně sekundárních vaskulitid při těchto nemocech. K určení
orgánové lokalizace a rozsahu postižení slouží vyšetření
nervového systému EMG, MR s angiografií a vyšetření
mozkomíšního moku. Ke stanovení definitivní diagnózy je
většinou potřeba provést biopsii postižené tkáně či orgánu.
Nejčastěji se indikuje biopsie spánkové tepny, kůže, nosní
sliznice a vedlejších dutin, ledvin, svalů, méně často
srdce, plic, periferních nervů nebo leptomening. U některých
typů vaskulitid stačí kvalitní zobrazovací metoda –
angiografie (klasická, digitální subtrakční, MR, CT)
a pozitronová emisní tomografie, významně může přispět
i ultrasonografické vyšetření tepen s barevným
dopplerovským vyšetřením.
B a C,
HIV a na hodnoty kryoglobulinů. Zásadní význam má
vyšetření ANCA a protilátky proti bazálním membránám
glomerulů, jejichž pozitivita často vede k diagnóze ANCA
asociovaných vaskulitid. Vyšetření antinukleárních protilátek,
autoprotilátek proti deoxyribonukleové kyselině,
ribonukleoproteinu, kardiolipinu slouží k diferenciální
diagnóze jiných běžnějších difuzních onemocnění pojiva,
případně sekundárních vaskulitid při těchto nemocech. K určení
orgánové lokalizace a rozsahu postižení slouží vyšetření
nervového systému EMG, MR s angiografií a vyšetření
mozkomíšního moku. Ke stanovení definitivní diagnózy je
většinou potřeba provést biopsii postižené tkáně či orgánu.
Nejčastěji se indikuje biopsie spánkové tepny, kůže, nosní
sliznice a vedlejších dutin, ledvin, svalů, méně často
srdce, plic, periferních nervů nebo leptomening. U některých
typů vaskulitid stačí kvalitní zobrazovací metoda –
angiografie (klasická, digitální subtrakční, MR, CT)
a pozitronová emisní tomografie, významně může přispět
i ultrasonografické vyšetření tepen s barevným
dopplerovským vyšetřením.
Závěr
Obecně platí, že vyšetření, stanovení diagnózy vaskulitidy a léčení těchto nemocných představuje pro většinu lékařů určitou „výzvu“, protože tyto vzácné choroby mohou vést k nevratnému poškození orgánů a v některých případech jsou i život ohrožující. Vždy je nutno nemocného s podezřením na vaskulitidu odeslat k revmatologovi. Podmínkou kvalitní péče je mezioborová interní spolupráce s kardiologem, angiologem, nefrologem, pneumologem a hematologem. Dále jsou často zapotřebí konzultace dermatologa, otorinolaryngologa, infekcionisty, neurologa, oftalmologa a specialistů na zobrazovací metody.
Seznam použité literatury
- [1] Bečvář R, Tesař V, Rychlík I. Vaskulitidy v klinické praxi – diagnostika a terapie. Praha: Medprint, 1994, 8.
- [2] Ansell BM, Bacon PA, Lie JT, et al. The vasculitides. Science and practice London: Chapman&Hall Medical 1996, 267–269.
- [3] Fessler BJ. Approach to the diagnosis of vasculitis in adult patients. In: Ball GV, Louis Bridge S Jr eds. Vasculitis, Oxford University Press, Oxford 2008, 277–284.
- [4] Alvarez‑Lafuente R, Fernández‑Gutiérrez B, Jover JA, et al. Human parvovirus B19, varicella zoster virus, and human herpes virus 6 in temporal artery biopsy specimens of patients with giant cell arteritis: analysis with quantitative real time polymerase chain reaction. Ann Rheum Dis 2005; 64: 780–782.
- [5] Hunder GG, Bloch DA, Michel BA, et al. The American College of Rheumatology 1990 revised criteria for the classification of giant cell arteritis. Arthritis Rheum 1990; 33: 1122–1128.
- [6] Pipitone N, Olivieri I, Salvarani C. Italian Society of Rheumatology Recommendations for the treatment of the primary large‑vessel vasculitis with biological agents. Clin Exp Rheumatol 2012; 30 (Suppl 70): S139–161.
- [7] Hoffman GS, Weyand CM, et al. Inflammatory diseases of blood vessels. New York: Marcel Dekker, Inc, 2002, 306.
- [8] Nikkari RA, Vainionpää R, Toivanen P, et al. Wegener‘s granulomatosis and parvovirus B19 infection. Arthritis Rheum 1995; 38: 1175–1176.
- [9] Csernok E, Holle J, Hellmich B, et al. Evaluation of capture ELISA for detection of neutrophil cytoplasmic antibodies directed against proteinase 3 in Wegeners granulomatosis: first results from a multicenter study. Rheumatology (Oxford) 2004; 43: 174–180.
- [10] Tarzi RM, Pusey CD. Current and future prospects in the management of granulomatosis with polyangiitis (Wegener‘s granulomatosis). Ther Clin Risk Manag 2014; 17: 279–293.
- [11] Calvino MC, Llorca J, Garcia‑Porrú AC, et al. Henoch‑Schönlein purpura in children in north western Spain: a 20‑year epidemiologic and clinical study. Medicine (Baltimore) 2001; 80: 279–290.
- [12] Bogdanović R. Henoch‑Schönlein purpura nephritis in children: risk factors, prevention and treatment. Acta Paediatr 2009; 98: 1882–1889.
- [13] Weiss PF, Feinstein JA, Luan X, et al. Effects of corticosteroid on Henoch‑Schönlein purpura: a systematic review. Pediatrics 2007; 120: 1079–1087.