Systémový lupus erythematodes
Souhrn:
Systémový lupus erythematodes (SLE) je autoimunitní systémové onemocnění postihující téměř všechny důležité orgány těla, zvláště pak kůži, klouby, kardiovaskulární systém, krvetvorbu, ledviny, plíce a centrální nervový systém. Je charakterizováno hyperaktivitou B lymfocytů, která vede k tvorbě autoprotilátek zaměřených proti orgánově nespecifickým antigenům. Dosud není jasné, zda se nejedná o syndrom, jehož společným jmenovatelem jsou uvedené projevy. Roční incidence nových případů je 1–10 nemocných/100 000 obyvatel. Laboratorně je pro SLE typická tvorba orgánově nespecifických protilátek namířených proti nukleárním, cytoplazmatickým i povrchovým antigenům vlastního těla. Jedná se o vysoce heterogenní chorobu, kterou lze dělit do řady klinicky a laboratorně definovaných podtypů. Průběh choroby je charakterizován střídáním remisí a exacerbací. Klinické příznaky jsou velmi pestré, nejčastějším prvním nálezem bývají artralgie až artritidy a typický motýlovitý exantém v obličeji, později se mohou přidat i další varianty postižení. Závažné jsou zejména formy s orgánovými projevy, kdy SLE může vyústit i do selhání postiženého orgánu a takto závažná forma choroby je spojena s významnou mortalitou. Systémový lupus erythematodes je častější u mladých žen a je značně rizikový i pro graviditu těchto pacientek. Vzhledem k rozmanité klinické symptomatologii je diagnostika SLE obtížná a ke stanovení diagnózy se používají tzv. klasifikační kritéria. Terapie SLE spočívá v kombinované imunosupresivní léčbě. Základní léčbu představují glukokortikoidy a v kombinaci s nimi jsou pak užívána jednotlivá další imunosupresiva, která jsou zvolena dle závažnosti jednotlivého postižení a dle aktivity nemoci. Zlepšení léčebných možností významně prodloužilo přežívání pacientů s tímto onemocněním.
Key words: systemic lupus erythematosus – clinical features – criteria – therapy – prognosis – pregnancy – immunosuppression – biological therapy.
Summary:
Systemic lupus erythematosus (SLE) is an autoimmune systemic disease affecting almost all important body organs, especially the skin, joints, cardiovascular system, hemopoiesis, kidneys, lungs, and central nervous system. It is characterized by hyperactivity of B‑lymphocytes, leading to production of autoantibodies against non‑organ‑specific antigens. It is not yet clear whether SLE does not represent a syndrome, the components of which just share the above manifestations. Annual incidence of SLE is 1–10 cases/100,000 inhabitants. Laboratory features of SLE include non‑organ‑specific autoantibodies against nuclear, cytoplasmic, and surface antigens of the patient’s body. SLE is a highly heterogeneous illness, which can be divided in many subtypes, defined by clinical and laboratory characteristics. Remissions and exacerbations occur in turns. Clinical features are very variable, arthralgias or arthritides and a typical facial “butterfly rash” usually manifesting first. SLE forms with organ involvement are the most severe since the respective organ failure may develop, increasing the mortality. Young females are affected by SLE more often than other people and SLE makes their possible pregnancies risky ones. The evaluation of SLE is difficult and requires the use of appropriate classification criteria. SLE is treated with combined immunosuppressive therapy based on glucocorticoids; individual other immunosuppressants are being added according to disease severity and activity. The survival of SLE patients has increased significantly since the spectrum of therapeutic options widened.
Úvod a definice
Systémový lupus erythematodes (SLE) je autoimunitní systémové onemocnění postihující téměř všechny důležité orgány těla, zvláště pak kůži, klouby, kardiovaskulární systém, ledviny, plíce a centrální nervový systém. Je charakterizované hyperaktivitou B buněk a nadprodukcí orgánově nespecifických autoprotilátek, které jsou namířeny proti nukleárním, cytoplazmatickým i povrchovým antigenům vlastního těla, z nichž mnohé se podílejí na tvorbě imunokomplexů. Jejich tkáňová či cévní depozita pak vedou k zánětlivému orgánovému postižení. Jedná se o vysoce heterogenní chorobu, kterou lze dělit do řady klinicky a laboratorně definovaných podtypů, jež se mohou s řadou dalších jednotek překrývat. Dosud není jasné, zda nejde o syndrom, jehož společným jmenovatelem jsou uvedené projevy [1].
Epidemiologie
Systémový lupus erythematodes se vyskytuje po celém světě. Prevalence SLE se liší podle jednotlivých populací a pohybuje se mezi 20–150 případy na 100 000 obyvatel. Roční incidence nových případů je kolem 1–10 nemocných na 100 000 obyvatel. Výskyt onemocnění je významně častější u žen ve fertilním věku a je asi desetkrát častější u žen než u mužů [2,3]. V rámci České republiky lze odhadovat počet nemocných se SLE na 6 000–10 000.
Etiopatogeneze
Specifická příčina tohoto onemocnění není dosud známa. Velká pozornost je věnována působení infekce, zvláště virové, buď jako spouštěcího mechanismu, nebo jako faktoru, který se aktivně podílí na průběhu a formě nemoci. Jiným etiologickým činitelem by mohly být zvýšené koncentrace některých pohlavních hormonů, zvláště estrogenu a prolaktinu, neboť zasahují významně do kvality imunitní odpovědi. Uvažovány jsou však i jiné etiopatogenetické faktory. Základem patogeneze tohoto onemocnění je tvorba autoprotilátek proti buněčným makromolekulám, jež vedou k poškození tkání životně důležitých orgánů [4,5]. Tato tvorba nadměrného množství autoprotilátek je podmíněna na prvním místě polyklonální aktivací B buněk, na druhém místě pak imunitní stimulací vyvolanou novými antigenními strukturami (autoantigeny). Autoprotilátkami zprostředkované poškození tkání pak vzniká na základě zánětlivé odpovědi vyvolané vzniklými imunitními komplexy (např. je tomu tak u glomerulonefritidy) nebo na základě buněčné dysfunkce, jež je opět zprostředkována autoprotilátkou (např. autoimunitní cytopenie).
Morfologicko histologický obraz se mění podle specifického složení postiženého orgánu, přičemž společným jmenovatelem je poškození cévní stěny – vaskulitida nebo nezánětlivá vaskulopatie.
Klinický obraz
Klinické příznaky jsou velmi mnohotvárné, takže si onemocnění vysloužilo označení „magna simulatrix“. Nemoc provázejí významné anamnestické údaje, mezi které patří nesnášenlivost slunečního záření, padání vlasů, ale též celkové křeče a mnohočetné bolestivé zduření mízních uzlin. Ze subjektivních obtíží vystupuje velmi často zvýšená únavnost, nadměrné pocení, artralgie a myalgie, z laboratorních nálezů pak vysoké hodnoty sedimentace erytrocytů, přetrvávající anémie, leukopenie či trombocytopenie. Nejčastějším prvním příznakem SLE jsou artralgie až artritidy, dále typický motýlovitý exantém na kůži obličeje či diskoidní změny kůže obličeje, nevůle, slabost a únavnost, horečka vyšší než 38 °C, někdy anorexie a úbytek hmotnosti.
Průběh onemocnění lze podle klinických zkušeností rozdělit do dvou období: tzv. časný průběh je ohraničen prvními pěti lety nemoci, poté následuje tzv. pozdní průběh, jehož doba trvání se neustále prodlužuje a již výrazně přesahuje 20leté přežívání.
V časném období choroby se nejčastěji objevují komplikace, které jsou důsledkem závažnosti a rozsahu příslušného orgánového postižení. Týká se to hlavně postižení ledvin, které se projevuje jejich předčasným akutním selháním, nebo postižení mozku ve formě akutní psychózy, cévní mozkové příhody, epileptických křečí nebo postižení plic ve formě akutní pneumonitidy či vzácně i plicní hemoragie. V popředí je v této době i autoprotilátková aktivita.
Pro pozdní období jsou charakteristické dlouhodobé důsledky nemoci, např. progredující renální insuficience nebo poškození mozku s neuropsychickými poruchami či s progresivní demencí. Zvláštní formou pozdního postižení je obraz osteonekrózy, která postihuje nejčastěji hlavici kyčelního kloubu, vyvíjí se postupně a je urychlována současnou terapií glukokortikoidy. S touto léčbou pravděpodobně souvisí předčasný vývoj tzv. akcelerované aterosklerózy s následnou pružníkovou hypertenzí. Jako důsledek dlouhodobé imunosupresivní léčby se v tomto období často objevují těžké infekční komplikace až septické stavy, které jsou nejčastější příčinou úmrtí na SLE. Rovněž se vzácněji může též vyvinout maligní nádorové onemocnění, většinou ve formě některé z hemoblastóz [6–9].
Kloubní projevy
Postižení kloubů polyartritidou je
velmi typickým projevem této nemoci, a to jak formou artralgií
a migrující subakutní polyartritidy, tak v podobě
těžších forem s vývojem deformit, jež imitují revm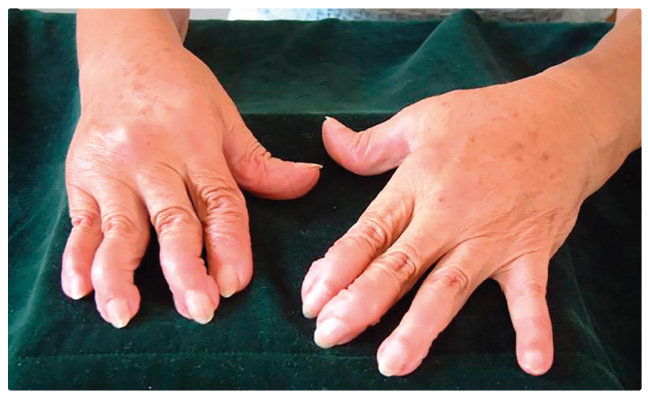 atoidní
artritidu. Avšak na rozdíl od této nemoci se udává
v literatuře též postižení distálních interfalangeálních
kloubů, neprovází je tak výrazná ranní ztuhlost a artritida
nevede k tvorbě kloubních ankylóz. Deformity zahrnují jak
subluxace kloubů, tak ulnární deviace prstů (obr. 1). V rentgenovém obraze nedochází k poškození
skeletu tvorbou erozí. Postižení kloubů bývá přítomno až
u 95 % všech nemocných [8].
atoidní
artritidu. Avšak na rozdíl od této nemoci se udává
v literatuře též postižení distálních interfalangeálních
kloubů, neprovází je tak výrazná ranní ztuhlost a artritida
nevede k tvorbě kloubních ankylóz. Deformity zahrnují jak
subluxace kloubů, tak ulnární deviace prstů (obr. 1). V rentgenovém obraze nedochází k poškození
skeletu tvorbou erozí. Postižení kloubů bývá přítomno až
u 95 % všech nemocných [8].
Kožní projevy
Kožní projevy jsou velice častým
příznakem choroby a vyskytují se až u 80 % nemocných.
Kožní projevy lze rozdělit na dvě základní formy: jednak
na nespecifické příznaky, jež vznikají na podkladě
předchozího poškození cév (chronické kožní ulcerace,
periferní gangrény, Raynaudův fenomén, teleangiektazie včetně
alopecie), jednak na specifické projevy. Ty mají tři základní
formy. Akutní kožní lupus (30–40 %) charakterizuje typický
obličejový „motýlovitý erytém“ na tvářích (obr. 2)
nebo se 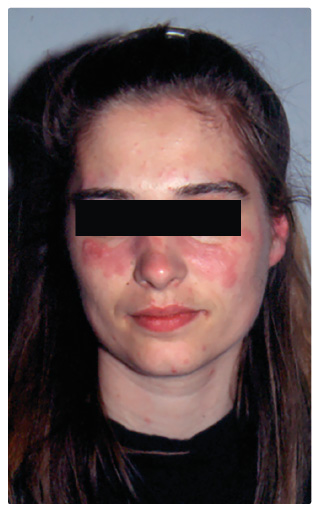 vyskytuje ve formě povrchového rozsevu morbiliformního
typu, jenž postihuje obličej, vlasatou část hlavy, šíji, horní
část hrudníku, ramena a dorzální část paží, rukou
i nohou. Často se tyto změny objeví po oslunění.
Subakutní kožní forma onemocnění (10–15 %) mívá
papulodeskvamózní ložiska bez jizvení, nejčastěji na kůži
šíje, horní části trupu a na končetinách. Ložiska
jsou spíše splývavá, někdy se podobají psoriáze. Chronický
kožní lupus (1–20 %) se nejčastěji objevuje ve formě
izolované diskoidní formy, pro niž je typický erytém, silná
adherující vrstva kožních folikulů s okolní
hyperpigmentací a tvorbou jizev. Nejvíce jsou postiženy
osvětlené části kůže a též boltce [10,11].
vyskytuje ve formě povrchového rozsevu morbiliformního
typu, jenž postihuje obličej, vlasatou část hlavy, šíji, horní
část hrudníku, ramena a dorzální část paží, rukou
i nohou. Často se tyto změny objeví po oslunění.
Subakutní kožní forma onemocnění (10–15 %) mívá
papulodeskvamózní ložiska bez jizvení, nejčastěji na kůži
šíje, horní části trupu a na končetinách. Ložiska
jsou spíše splývavá, někdy se podobají psoriáze. Chronický
kožní lupus (1–20 %) se nejčastěji objevuje ve formě
izolované diskoidní formy, pro niž je typický erytém, silná
adherující vrstva kožních folikulů s okolní
hyperpigmentací a tvorbou jizev. Nejvíce jsou postiženy
osvětlené části kůže a též boltce [10,11].
Kardiovaskulární projevy
Postižení srdce patří mezi časté příznaky tohoto onemocnění (30–50 %). Pojem lupusová karditida zahrnuje perikarditidu, myokarditidu, Libmanovu–Sacksovu endokarditidu a také vaskulitické postižení koronárních cév. Nejčastějším projevem srdečního postižení je však až u 60 % případů perikarditida s perikardiálním výpotkem, obvykle s minimálními klinickými projevy, vzácnou komplikací pak může být i srdeční tamponáda či tvorba perikardiálních srůstů. Závažným postižením je endokarditida, jež ovšem bývá někdy klinicky němá a lze ji zjistit pouze echokardiograficky při jícnové echokardiografii nebo až při autopsii. Je však nutno počítat s možností rozvoje nasedající sekundární bakteriální endokarditidy.
Postižení koronárních tepen typu vaskulitidy nebo vaskulopatie je méně časté a bývá kombinováno s projevy tzv. akcelerované aterosklerózy, jejíž výskyt se dává mimo jiné do souvislosti s dlouhodobou léčbou glukokortikoidy. Vaskulopatie pak bývá vyvolána antifosfolipidovými autoprotilátkami. Plicní hypertenze se u SLE vyskytuje v důsledku vaskulopatie či vaskulitidy méně často než například u sklerodermie, je udávána asi ve 4–11 % případů, ale vždy je vysoce závažnou komplikací. Asi 13 % nemocných se SLE má manifestní projevy akcelerované aterosklerózy (angina pectoris, infarkt myokardu, projevy ischemie v důsledku postižení periferních cév). Počet úmrtí v souvislosti s důsledky kardiovaskulárních komplikací pravděpodobně dokonce převyšuje počet úmrtí v souvislosti s vlastní aktivitou choroby [8,9]. Riziko akcelerace aterosklerózy zvyšuje také léčba SLE glukokortikoidy, především v důsledku indukované hyperlipoproteinemie.
Plicní projevy
Pleuritida patří vedle perikarditidy mezi nejčastější postižení serózních blan. Také v pleurálním výpotku lze prokázat přítomnost autoprotilátek a imunitních komplexů. Plicní parenchym může být postižen v několika formách. Zřídka jde o akutní lupusovou pneumonitidu nebo krvácení do alveolů. Projevují se náhlou a těžkou respirační insuficiencí, jež ohrožuje nemocného na životě. Druhou formou je chronická difuzní intersticiální pneumonitida, která se postupně vyvíjí a omezuje výkonnost nemocného chronickou dušností. Není dosud vyjasněno, zda je následkem akutní pneumonitidy, nebo nezávislým projevem základního onemocnění. Vážnou komplikací je postupně vznikající plicní hypertenze, pravděpodobně na základě cévních okluzí různého původu, jako jsou vaskulitida či vaskulopatie, trombóza či intersticiální pneumonitida [7,9].
Neuropsychiatrický SLE
Postižení centrální nervové soustavy se projevuje velmi různorodými příznaky, jejichž přesná charakteristika a diagnostika bývá značně obtížná. Je to převážně postižení mozku, případně míchy, jehož podstata však není dosud zcela vyjasněna. Podkladem těchto změn je nezánětlivé postižení cév, tedy vaskulopatie s následnou tvorbou trombů a mikroinfarktových ložisek v mozkové tkáni. Předpokládá se zde i přímý vliv antifosfolipidových autoprotilátek. Jen vzácně jsou histologicky prokázány projevy vaskulitidy. Postupně se zjišťuje, že postižení mozku je daleko častější – dochází k němu až u 60 % nemocných a představuje jednu z předních příčin úmrtí u tohoto onemocnění. Neuropsychiatrický SLE může mít projevy jak neurologické, tak i psychiatrické. K neurologickým projevům patří epiparoxysmy různého typu, mozkové příhody, pohybové poruchy, bolesti hlavy nemigrenózního typu, transverzální myelitida a centrální a periferní neuropatie. Mezi projevy psychiatrického SLE je řazen organický mozkový syndrom se změnami chování až s vývojem progresivní demence, dále pak psychózy, psychoneurózy a neurokognitivní dysfunkce [12,13].
Renální projevy
Onemocnění ledvin je u SLE vždy vážným projevem imunopatologického procesu a zůstává stále jednou z nejčastějších příčin smrti. Moderní způsoby léčení už výrazně zvýšily pravděpodobnost přežití těchto nemocných z původních 5 let pro 90 % nemocných až na 15 let. Klinický syndrom ledvinového postižení – glomerulonefritidy – může být až v 10 % prvním příznakem choroby. Výsledky renálních biopsií ukázaly, že každý nemocný se SLE má určité glomerulární abnormality, jež však nemusejí progredovat. Poměr ledvinového postižení mezi oběma pohlavími je podobný jako u celé nemoci, tj. 10–12 žen k jednomu muži. Nejčastěji se objevuje ve druhé až třetí dekádě života. V podstatě jde o imunokomplexové onemocnění lokalizované do glomerulu [8,9].
Klinicky je diagnóza ledvinového poškození vázána na vývoj tzv. aktivního močového syndromu, který spočívá v přítomnosti proteinurie a výskytu nadměrného množství erytrocytů a převážně granulárních válců v močovém sedimentu. Za hranici patologických hodnot je pokládána proteinurie v množství 0,5 g/24 hodin trvající alespoň 3–6 měsíců. Není dosud objasněno, kteří nemocní přecházejí ze stadia klinicky němého postižení ledvin do stadia aktivního, jež se projevuje proteinurií. K základním příznakům ledvinového postižení patří tedy proteinurie, tzv. aktivní močový sediment (nález erytrocytů nebo válců) a hypertenze, dále známky renální insuficience se zvýšenými hodnotami urey a kreatininu v séru a s poklesem glomerulární filtrace. Jednotlivé typy ledvinového postižení podle Světové zdravotnické organizace (WHO) mají pak různé klinické známky a různou prognózu vývoje až do konečného stadia ireverzibilního renálního selhání.
Hematologické projevy
Často je u SLE přítomna anémie, buď v důsledku snížené tvorby červených krvinek, nebo následkem imunopatologické hemolýzy. Mezi hematologické projevy patří rovněž leukopenie, lymfopenie a trombocytopenie, jež se rozvíjejí v aktivní fázi onemocnění vlivem autoprotilátek. Zajímavou hematologickou abnormalitou je přítomnost autoprotilátky lupus antikoagulans, jež je jednou z tzv. antifosfolipidových protilátek. Její přítomnost se mimo jiné speciální testy projevuje i prodlouženým aktivovaným parciálním tromboplastinovým časem (activated partial tromboplastine time, aPTT). Dalším typem antifosfolipidových autoprotilátek jsou protilátky proti antikardiolipinům (anticardiolipin antibodies, ACLA) a protilátky proti β2 glykoproteinu [7,9].
Další laboratorní projevy
Pro hodnocení laboratorních znaků rozlišujeme jednak nespecifické laboratorní příznaky zánětu, tj. sedimentace erytrocytů, anémie, zvýšené hodnoty C reaktivního proteinu, fibrinogenu a α1 antitrypsinu a snížení hodnoty ceruloplazminu, jednak laboratorní příznaky související s orgánovým postižením, např. ledvin, plic, srdce atd. Velmi důležitou úlohu hrají specifické laboratorní příznaky, jež zahrnují imunologické abnormality provázející toto onemocnění; na prvním místě to jsou cirkulující antinukleární autoprotilátky (ANA) a jejich specifické podtypy a dále protilátky proti extrahovatelným nukleárním antigenům (ENA) s příslušnými podtypy. Největší diagnostický význam má přítomnost protilátky proti nativní DNA (anti dsDNA), zvláště pro poškození ledvin. Podobný význam je nutno přičíst též specifické autoprotilátce ze skupiny ENA zvané anti Sm. Významnou součástí laboratorního průkazu onemocnění a jeho aktivity je stanovení komplementu, a to jak jeho celkového množství (CH50), tak jeho některých významných složek, zvláště C3 a C4, jejichž hodnoty jsou v aktivním stadiu onemocnění významně sníženy. Součástí laboratorního vyšetření je pak i stanovení koncentrace imunoglobulinů podle jednotlivých tříd a stanovení koncentrace imunitních cirkulujících komplexů. Z těchto testů lze pak složit určitý soubor, podle něhož můžeme předpovědět hrozící nebo stávající exacerbaci onemocnění [7–9].
Diagnostika a diferenciální diagnostika
Diagnostika choroby se opírá o správnou interpretaci řady anamnestických, klinických, laboratorních a paraklinických nálezů. Pro diagnózu SLE neexistuje žádný zlatý standard, nález, příznak či vyšetření, které by s velmi vysokou pravděpodobností diagnózu choroby potvrdily.
Ke stanovení diagnózy se
ustálilo používání tzv. diagnostických kritérií
podle Americké revmatologické společnosti (American
College of Rheumatology, ACR) z roku 1982 (tab. 1). V současné době jsou k dispozici ještě
novější klasifikační kritéria SLICC (Systemic Lupus
International Collaborating Clinic), kte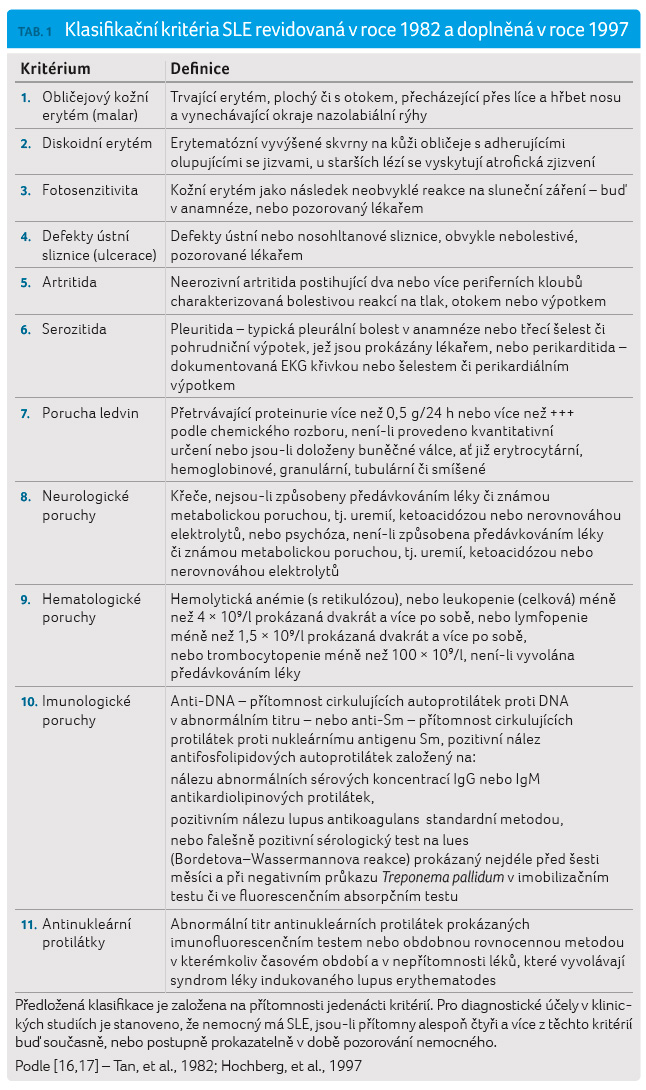 rá byla validována
a publikována v roce 2012 (tab. 2). Skládají se ze dvou částí: z klinických
a laboratorních kritérií. Oproti kritériím ACR jsou
rozšířena o neurologické, hematologické a imunologické
nálezy. V současnosti lze pro klasifikaci SLE použít oba dva
systémy [14–18].
rá byla validována
a publikována v roce 2012 (tab. 2). Skládají se ze dvou částí: z klinických
a laboratorních kritérií. Oproti kritériím ACR jsou
rozšířena o neurologické, hematologické a imunologické
nálezy. V současnosti lze pro klasifikaci SLE použít oba dva
systémy [14–18].
Rozpozn ání SLE je někdy velmi
nesnadné, neboť klinická symptomatologie je velmi často velice
rozmanitá. Nemoc obvykle začíná nespecifickými celkovými
příznaky choroby, jako je přetrvávající zvýšená teplota nebo
vysoká horečka bez zřejmých infekčních příčin spojená se
značnou celkovou schváceností a s velkou únavou. V této
době je diagnostika velmi obtížná a na SLE je obvykle
pomýšleno až v případě vývoje specifičtějších
příznaků, jako jsou postižení kůže, kloubů a zejména
přítomnost specifických imunologických abnormalit, resp.
autoprotilátek. Někdy se ale SLE může manifestovat rovnou
závažným orgánovým postižením.
ání SLE je někdy velmi
nesnadné, neboť klinická symptomatologie je velmi často velice
rozmanitá. Nemoc obvykle začíná nespecifickými celkovými
příznaky choroby, jako je přetrvávající zvýšená teplota nebo
vysoká horečka bez zřejmých infekčních příčin spojená se
značnou celkovou schváceností a s velkou únavou. V této
době je diagnostika velmi obtížná a na SLE je obvykle
pomýšleno až v případě vývoje specifičtějších
příznaků, jako jsou postižení kůže, kloubů a zejména
přítomnost specifických imunologických abnormalit, resp.
autoprotilátek. Někdy se ale SLE může manifestovat rovnou
závažným orgánovým postižením.
Diferenciálně diagnosticky je nejčastěji nutno vyloučit jiná horečnatá celková onemocnění, zejména infekci, bakteriální endokarditidu až i sepsi neznámé etiologie, dále onemocnění nádorové, zejména hemoblastózy, a rovněž i jiná revmatologická zánětlivá systémová onemocnění [7,8,15].
Prognóza
Dlouhodobé přežívání nemocných se SLE je závislé především na rozsahu a závažnosti orgánové manifestace SLE, tedy specifického postižení ledvin, mozku, plic či srdce. V posledních 20–30 letech došlo k významnému prodloužení přežívání nemocných se SLE. K této změně přispěl zejména výrazný posun ve znalostech imunopatologických mechanismů nemoci, což vede jednak ke zlepšení a urychlení diagnostiky, jednak k využití zcela nových terapeutických možností, v posledních letech i s vývojem tzv. biologických léků. Terapeutické možnosti výrazně zlepšila i velmi dobrá dostupnost renální dialýzy a transplantace ledvin [7,19].
Gravidita
Systémový lupus erythematodes postihuje zejména mladé ženy ve fertilním věku. Pokud žena se SLE otěhotní, podaří se jí to obyčejně v období nízké aktivity základního onemocnění, kdy není fertilita poškozena. Nicméně těhotenství je nutno každopádně pokládat za dosti rizikové, protože na jedné straně gravidita ovlivňuje průběh základního onemocnění, např. nebezpečím vyvolání exacerbace SLE, na druhé straně choroba sama ovlivňuje vlastní průběh těhotenství. Rizikovost gravidity u nemocných žen se SLE vyplývá ze samotné podstaty onemocnění SLE, které je vázáno zejména k ženskému pohlaví, vzniká nejčastěji ve fertilním věku a jeho vznik i průběh jsou pravděpodobně významně závislé na estrogenních hormonech. Onemocnění má v průběhu gravidity či v laktaci tendenci k relapsům a není vzácné, že se v průběhu gravidity manifestuje úplně poprvé. Mezi nejčastější komplikace gravidity u SLE patří možnost exacerbace onemocnění, dále vznik tzv. lupus neonatorum, větší četnost spontánních potratů a riziko vzniku preeklampsie [20–24].
Exacerbace
Exacerbace SLE může vzniknout rovnoměrně v průběhu všech trimestrů gravidity a v období krátce po porodu. V minulosti byla popisována až u 50 % gravidních žen se SLE, avšak v posledních 20 letech její výskyt významně klesá. U pacientek, u nichž bylo onemocnění v době před početím v remisi, se frekvence výskytu vzplanutí nemoci v průběhu gravidity přibližuje k počtu exacerbací u žen, které gravidní nejsou. Nejčastější formou relapsu SLE bývají nefritida nebo kožní a kloubní projevy. U žen s lupusovou nefritidou hrozí v průběhu těhotenství vyšší riziko ztráty plodu, které je udáváno až do 75 %, a dále větší riziko exacerbace nemoci ve formě jednak progrese nefropatie, jednak vzniku jiných orgánových projevů. V případě, že v době před početím byla nefritida alespoň šest měsíců v remisi, je frekvence exacerbace v době gravidity udávána od 7 % do 33 %. U žen, jež před početím trpěly aktivní nefritidou, je výskyt vzplanutí vysoký (popisován u 61–67 % případů). Toto riziko ještě stoupá u těch žen, které měly v době před početím hypertenzi.
Preeklampsie a ztráta plodu
Závažnou komplikací průběhu gravidity může být vznik preeklampsie, která je nejen tíživým zdravotním, ale i diagnostickým problémem, neboť bývá obtížné odlišit ji od vzplanutí lupusové nefritidy. Preeklampsie je popisována u 13 % těhotných žen a vyskytuje se zejména u nemocných s přítomností antifosfolipidových protilátek nebo s anamnézou lupusové nefritidy, hypertenze a diabetu [21,23].
U žen se SLE je rovněž vyšší riziko ztráty plodu, které je udáváno mezi 20–30 %, některými autory až v 50 %. Mezi významné rizikové faktory pro ztrátu plodu patří přítomnost antifosfolipidových protilátek a projevy vysoké aktivity SLE, zejména pak vysoký titr protilátek proti dvojspirálové DNA, nízká hodnota komplementu a lupusová nefritida [25]. Takto nemocné ženy totiž nejsou často schopny plod donosit a ztrácejí jej opakovaným potrácením nejčastěji ve druhém nebo na začátku třetího trimestru. Za příčinu tohoto jevu je pokládána přítomnost a působení antifosfolipidových autoprotilátek, a to jak lupus antikoagulans, tak antikardiolipinových protilátek a protilátek proti β2 glykoproteinu. Předpokládá se, že tyto protilátky vyvolávají trombózu cév placenty a tím negativně ovlivňují přívod kyslíku a výživy k plodu, což vede k jeho předčasnému zániku. Tomu lze předcházet včasně nasazenou antiagregační a antikoagulační léčbou, která je u rizikových žen podávána téměř po celou dobu gravidity [25–29].
Lupus neonatorum
Dalším rizikem pro plod je přítomnost cirkulujících autoprotilátek typu anti Ro a anti La v organismu matky, které mohou vyvolat u plodu vznik lupus neonatorum, jehož nejzávažnějším, i když vzácným projevem je kompletní srdeční atrio ventrikulární (A V) blok. Lupus neonatorum se však vyskytuje nejen u pacientek se SLE, ale i u žen s jinými systémovými chorobami, v jejichž autoprotilátkovém spektru nacházíme pozitivitu zmíněných autoprotilátek anti Ro a anti La, které jsou transplacentárně přeneseny do organismu plodu.
Terapeuticky jej lze ovlivnit glukokortikoidy s nízkou molekulovou hmotností, které pronikají placentou (např. dexametazon), ale efekt není jistý a jeho podmínkou je naprosto včasné zahájení terapie bezprostředně po vzniku blokády. Její časná detekce je pak podmíněna častým prováděním fetální echokardiografie podle doporučených schémat [23–34].
Významnou roli v graviditě hraje rovněž typ terapie, kterou je žena před graviditou i v jejím průběhu léčena a která může mít negativní vliv na plod. Naprostá většina pacientek se SLE je léčena glukokortikoidy, jež jsou v obvyklých nižších dávkách bezpečné a ani eventuální přechodné zvýšení jejich dávky v případě nutnosti nepředstavuje vysoké riziko. Nemocné s orgánovou manifestací SLE ale bývají léčeny i kombinovanou imunosupresivní léčbou s cytotoxickými léky a ty jsou pro graviditu zásadně kontraindikovány. Vysazení takovéto léčby není ovšem vždy možné a bezpečné, a tudíž je nutno u každé nemocné se SLE dle možností graviditu řádně naplánovat na vhodnou dobu, kdy je nemoc v remisi, nebo alespoň stabilizovaná při terapii, jež je i při graviditě bezpečná. Vysoce riziková je z hlediska gravidity i laktace zejména terapie cyklofosfamidem, při níž je nutno mimo jiné před koncepcí preventivně zajistit ochranu vajíčka, aby nevznikla ireverzibilní amenorea. Nejvhodnější formou takovéto prevence je hormonální antikoncepce nebo zmrazení vajíčka. Rovněž terapie cyklosporinem A není v graviditě vhodná, ovšem v případě ohrožení života gravidní nemocné se SLE ji lze použít. Terapie antimalariky je na základě novějších analýz a doporučení považována za velmi vhodnou, jelikož má řadu pozitivních vlivů, a obecně se připouští, že riziko relapsu SLE v graviditě a jeho důsledky výrazně převýší možné riziko případných nežádoucích účinků terapie na plod [22,30–34].
Léky indukovaný lupus erythematodes
Jedná se o klinický syndrom, který klinicky i laboratorně imituje idiopatický SLE. Je známo více než 30 léčiv, která mohou toto onemocnění vyvolat [8,9]. Nejčastěji mezi ně patří antiarytmika (prokainamid), antihypertenziva (hydralazin, metyldopa), dále chlorpromazin, penicilamin, isoniazid, z přípravků využívaných v samotné revmatologii pak sulfasalazin a z novějších léčiv blokátory tumor nekrotizujícího faktoru alfa. Léky indukovaný SLE postihuje ve stejném poměru ženy i muže, čímž se liší od samotného SLE, a jeho projevy postupně regredují od okamžiku, kdy je ukončena expozice vyvolávajícímu přípravku. V etiopatogenezi tohoto syndromu hraje roli mimo jiné i určitá genetická dispozice mající vztah k rychlosti acetylace, a tím i k odbourávání rizikových látek v játrech.
Terapie systémového lupus erythematodes
Terapii SLE lze rozdělit na tři roviny – předně to je specifická léčba mířená proti tvorbě autoprotilátek, která vede k útlumu působení autoreaktivních buněk. Na druhém místě pak jsou to léky symptomatické (analgetika a antipyretika), jež současně působí protizánětlivě, např. nesteroidní antirevmatika včetně kyseliny acetylsalicylové. A nakonec jsou to léky, jejichž podávání je vynuceno postižením funkce jednotlivých důležitých orgánů, jako jsou ledviny, srdce, plíce atd. Cílem léčby je minimalizovat celkové projevy zánětu a předejít orgánovému postižení nebo zabrzdit jeho progresi [8].
K lékům první roviny počítáme na předním místě glukokortikoidy, jež vedle nespecifického účinku protizánětlivého mají též významný a fyziologicky nejšetrnější efekt imunosupresivní. Proto u aktivních případů SLE se známkami postižení životně důležitých orgánů podáváme předně účinnou dávku glukokortikoidů v dávce 40–60 mg prednisonu nebo metylprednisolonu denně. Výše dávky závisí zejména na tom, kdy získáme kontrolu nad onemocněním. U méně závažných případů je dávka glukokortikoidů pochopitelně výrazně nižší.
Při těžkých stavech základního onemocnění se závažnými orgánovými projevy, zejména s postižením ledvin a CNS, se v úvodu léčby osvědčuje tzv. pulzní typ léčby s aplikací vysokých dávek (500–1 000 mg) metylprednisolonu v jednorázové infuzi opakované ve shodných intervalech ob den podle závažnosti postižení alespoň 3× po sobě. Nemocným se SLE, pokud se nenacházejí v úplné dlouhodobé remisi, většinou musí být podávána udržovací dlouhodobá léčba v denní dávce 5–10 mg prednisonu. Léčba SLE spočívá ve většině případů v kombinované imunosupresi, kdy základním lékem jsou glukokortikoidy a do kombinace k nim jsou přidána další imunosupresiva, jejichž volba je individuální a záleží na řadě okolností, a to zejména na aktivitě a závažnosti základního onemocnění SLE, na polymorbiditě pacienta a i na tom, zda žena plánuje těhotenství.
U nemocných s převážně kožní a kloubní formou nebo u nemocných s nižší aktivitou onemocnění je možno podávat antimalarika, nejlépe hydroxychlorochin dlouhodobě v dávce 200 mg denně za kontroly oftalmologem nebo při neúspěchu této terapie pak metotrexát v dávce do 25 mg týdně.
U nemocných trpících SLE
se závažným postižením životně důležitých orgánů nebo
s projevy vysoké autoprotilátkové aktivity podáváme
do kombinace s glukokortikoidy zejména cytotoxické
látky s vysokým imunosupresivním účinkem. Je to
nejčastěji azathioprin, mykofenolát mofetil nebo cyklosporin A
v perorální formě. U těžkých forem onemocnění je
indikován cyklofosfamid,
přičemž nejvhodnější formu jeho aplikace představuje série
intravenózních pulzů. Tyto léky vedou k poklesu aktivi ty
nemoci až k navození a udržení remise a umožňují
i snížit denní dávku glukokortikoidů (tab. 3) [35].
ty
nemoci až k navození a udržení remise a umožňují
i snížit denní dávku glukokortikoidů (tab. 3) [35].
U závažných forem lupusové nefritidy (třídy IV nebo V podle WHO) nebo u závažné formy neuropsychiatrického SLE se doporučuje pulzní forma glukokortikoidů, případně synchronizovaná s plazmaferézou nebo s imunoadsorpčními postupy a intermitentní pulzní podávání cyklofosfamidu i.v. v celkové dávce 400–800 mg (10 mg/kg) v opakovaných časových intervalech od tří týdnů až po tři měsíce v trvání od půl roku do jednoho roku, vzácně déle. Tato forma se jeví jako výhodnější s ohledem na výskyt nežádoucích toxických projevů. Při dlouhodobém užití těchto léků je nutno sledovat možnost výskytu oportunní celkové infekce nebo při perorálním podávání cyklofosfamidu i hemoragické cystitidy. Je nutno brát v úvahu i možné projevy teratogenity a pozdní malignity.
Dalším vysoce účinným imunosupresivem je nověji i mykofenolát mofetil, který je využíván zejména v léčbě lupusové nefritidy. Velice účinnou variantou v léčení závažných forem SLE je užití imunomodulačního léčiva cyklosporinu A v relativně nízkých dávkách, což je efektivní zvláště při postižení ledvin s vývojem do nefrotického syndromu. U těžkých a na standardní léčbu refrakterních případů lze podat cyklosporin A v dávce do 5 mg/kg/den nebo konečně vysoké dávky koncentrovaných imunoglobulinů. Limitující pro tuto terapii jsou ale její nežádoucí účinky, zejména vyvolání hypertenze a projevy nefrotoxicity.
Léčba intravenózními gamaglobuliny
je stále předmětem diskuse. Na zahájení léčby u pacientů
s vysokou aktivitou onemocnění lze ve vzácných
případech úspěšně použít i plazmaferézu. Významný
posun doznalo léčení terminální ledvinové nedostatečnosti
v programu chronické intermitentní hemodialýzy s perspektivou
úspěšné transplantace ledviny (tab. 4) [14].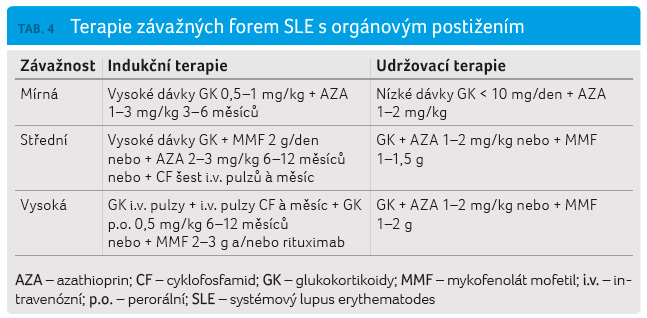
Efektivní léčba vyžaduje přesné stanovení aktivity i závažnosti choroby. Aktivita choroby se vztahuje většinou ke stupni zánětu, zatímco závažnost choroby závisí na typu orgánového postižení a dysfunkce a na relativním významu postiženého orgánu či tkáně. Aktivita choroby je hodnocena kombinací anamnézy, klinického vyšetření, orgánově specifických testů a sérologických biomarkerů. Kvantifikaci aktivity choroby umožňují validované skórovací systémy aktivity (SLEDAI, BILAG atd.). Pro běžnou klinickou praxi je nejvhodnější systém SLEDAI (SLE Diasease Activity Index) [15].
Současná terapie SLE umožní 15leté přežití v 85 % případů, což je výrazný pokrok oproti pětiletému přežití dosahovanému u 50 % nemocných v polovině minulého století. Kromě uvedené tradiční imunosupresivní terapie SLE se v poslední době začíná objevovat snaha o cílenou léčbu zaměřenou na ovlivnění patofyziologických mechanismů vzniku a akcelerace onemocnění.
Dalšího zlepšení prognózy nemocných se SLE bude možné dosáhnout díky cílené terapii vycházející z poznatků studia imunopatogenetických mechanismů onemocnění. V posledních letech probíhá intenzivní vývoj a testování molekul působících na úrovni kostimulace T lymfocytů antigen prezentujícmi buňkami, léků působících depleci B lymfocytů pomocí protilátek proti jejich povrchovým molekulám (anti CD20 a anti CD22) nebo omezujících jejich vyzrávání a přežití inhibicí faktoru stimulujícího B lymfocyty (BAFF, BLyS). Testovány jsou molekuly, které interferují s aktivací komplementu, s cytokiny, s interferony a s funkcí toll like receptorů, a léky, které mají navodit toleranci k autoantigenům [36,37]. Některé léky mohou navodit remisi, jiné prodloužit její trvání. Další výzkum bude nejspíše zaměřen na testování vhodných kombinací a časování dle fází onemocnění [38–40].
Biologická léčba u SLE
Klíčovou úlohu v patogenezi SLE hrají B lymfocyty. Jejich význam spočívá v jejich schopnosti fungovat jako antigen prezentující buňky schopné produkovat cytokiny a aktivovat buněčnou zánětlivou odpověď prostřednictvím aktivace T lymfocytů. Při vzniku a progresi choroby se dále uplatňují autoreaktivní B lymfocyty, a to svou schopností produkovat autoprotilátky s následnou aktivací komplementového systému vedoucí ke vzniku imunokomplexů, které v konečné fázi způsobují poškození tkání. Vzhledem k uvedené významné roli B lymfocytů v patogenezi SLE je biologická terapie této nemoci zaměřena právě na ovlivnění těchto buněk.
Belimumab
Prvním a doposud jediným oficiálně registrovaným biologickým přípravkem je belimumab – plně humánní rekombinantní IgG1λ monoklonální protilátka, která blokuje vazbu solubilního stimulátoru B lymfocytů (B lymphocyte stimulator, BLyS) na B lymfocyty. V České republice byl tento lék registrován již v roce 2011 a nyní se dlouhodobě projednává jeho indikace a úhrada.
BLyS nebo také BAFF (B cell activating factor, B lymfocyty aktivující faktor) je jedním z klíčových cytokinů ovlivňujících maturaci a přežívání různých diferenciačních stadií B lymfocytů, včetně autoreaktivních klonů. Exprimuje se na povrchu buněk, kde dochází k jeho štěpení na solubilní proteinový fragment. Zvýšené koncentrace solubilního BLyS a zároveň jeho zvýšená buněčná exprese byly pozorovány u pacientů s autoimunitními onemocněními včetně SLE. Blokáda vazby solubilní části BLyS na jeho receptory spojená s inhibicí jeho aktivity se ukázala jako možný terapeutický přístup v léčbě SLE. Tato selektivní vazba blokuje CD20+ B lymfocyty a plazmatické buňky s krátkým biologickým poločasem [36]. Byla prokázána korelace s koncentracemi BLyS a s aktivitou SLE, nicméně přesný patogenetický význam BLyS u SLE zůstává dosud nejasný.
Belimumab se neváže přímo
na B lymfocyty, ale prostřednictvím vazby na solubilní
BLyS a vytvořením komplexu s ním blokuje vazbu BLyS
na jeho receptory na B lymfocytech; tím dojde
ke snížení aktivace, diferenciace a přežívání
B lymfocytů (obr. 3).
To se následně projeví i s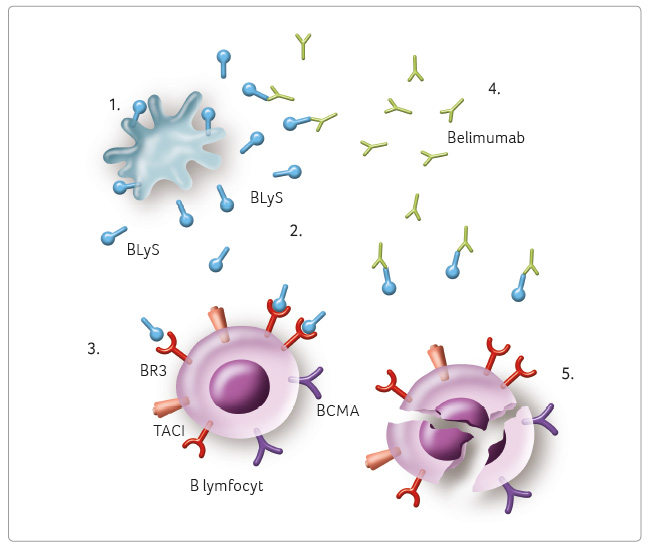 nížením tvorby autoprotilátek
a aktivace T lymfocytů.
nížením tvorby autoprotilátek
a aktivace T lymfocytů.
U pacientů léčených belimumabem byly v klinických studiích zjištěny výrazně snížené hodnoty cirkulujících C19+, CD20+ aktivovaných B lymfocytů. Při dlouhodobém klinickém sledování byly pozorovány statisticky signifikantně snížené koncentrace B lymfocytů v 52. týdnu léčby (p = 0,008). Studie prokázaly, že belimumab ovlivňuje nejdříve pokles počtu nově aktivovaných B lymfocytů (ve 12. a 14. týdnu; p < 0,05), pak až pokles počtu plazmatických buněk a paměťových buněk (v 76. týdnu; p < 0,01 a p < 0,05). Byly zaznamenány i změny dalších imunologických parametrů, u pacientů s hypergamaglobulinemií došlo k normalizaci koncentrací IgG až ve 49 % do 52. týdne léčby. Normalizace sníženého komplementu nastala u 44 % pacientů léčených belimumabem oproti 19 % v placebové skupině. Rovněž bylo pozorováno vymizení protilátek proti dvouvláknové DNA (anti dsDNA) u 16 % pacientů v 52. týdnu ve srovnání se 7 % pacientů dostávajících placebo [37–39].
Belimumab je lék s relativně dobrým bezpečnostním profilem a nejsou známy žádné významné nežádoucí účinky léčby, které by jeho aplikaci omezily [37]. Podávání belimumabu je započato indukční dávkou 10 mg/kg ve dnech 0, 14 a 28, poté je následováno udržovací terapií, kdy je infuze podávána ve čtyřtýdenních intervalech. Pokud nedošlo ke zlepšení po šesti měsících podávání, je nutné léčbu přerušit. Podávání belimumabu není doporučováno u pacientů se závažnou aktivní lupusovou nefritidou, s postižením CNS, s infekcí HIV, s hepatitidou B a C, s hypogamaglobulinemií a u pacientů po orgánové transplantaci [35,39].
Rituximab
Rituximab nemá ve své indikaci zařazen SLE, ale řada studií i dlouhodobých celosvětových zkušeností prokazuje jeho účinnost u této nemoci, takže je možno jej podávat i mimo indikační spektrum na základě schválení revizním lékařem.
Rituximab je chimérická IgG1 monoklonální protilátka složená z myší a lidské komponenty, která je namířena proti CD20. Antigen CD20 je exprimován na B lymfocytech, ale jeho exprese nebyla popsána na kmenových a plazmatických buňkách či jiných tkáních. Rituximab vede k lýze CD20+ buněk prostřednictvím vazby Fab části na CD20 a jeho Fc fragment pak zodpovídá za aktivaci celého imunitního procesu. Rituximab svou vazbou na CD20 je schopen mimo výše zmíněné vyvolat apoptózu B lymfocytů. Studie s rituximabem prováděné in vitro prokázaly výraznou depleci a zánik B lymfocytů po podání této látky. Rituximab je s úspěchem využíván v terapii lymfomů, polyangiitidy s granulomatózou a v terapii revmatoidní artritidy. V terapii SLE je jeho indikace omezena na obzvláště závažné případy orgánového postižení, jako je lupusová nefritida po selhání konvenční terapie [35].
Výzkum dalších biologických léčiv
Ve fázi výzkumu biologické terapie SLE se nacházejí i další molekuly a je posuzováno i uplatnění již známých biologických léčiv, která jsou doposud využívána v terapii jiných autoimunitních chorob. V současnosti probíhají klinické studie fáze II–III u řady těchto dalších biologických léků. Efekt přípravku epratuzumab (anti CD22) a abetimus (blokátor specifických B buněk a anti dsDNA protilátek) nebyl ve studiích prokázán a jejich výzkumné sledování bylo zastaveno [40,41]. Naopak další klinické studie probíhají (atacicept, abatacept, ocrelizumab, tocilizumab, ofatumumab, rapamycin, anakinra) a jejich výsledky teprve zhodnotí případný přínos těchto přípravků pro léčbu choroby [42]. Dále je experimentálně zkoušen i eculizumab, infliximab, spliceozomální peptid, anti B7RP 1 monoklonální protilátka, anti IL10 monoklonální protilátka, anti CD40 monokolonální protilátka, anti IFNα monoklonální protilátka. Potenciální terapeutický efekt je zvažován i u bortezomibu, terapie anti IL21 a Syk (inhibitor tyrozinkinázy).
Seznam použité literatury
- [1] Salmon JE, Pricip LD, Agati V. Immunopathology of systemic lupus erythematosus. In Hochberg, MC., et al. Rheumatology. Elsevier, 2008, p. 1217–1237.
- [2] Danchenko N, Satia JA, Anthony MS. Epidemiology of systemic lupus erythematosus: a comparison of worldwide disease burden. Lupus 2006; 15: 308–318.
- [3] Chakravarty EF, Bush TM, Manzi S, et al. Prevalence of adult systemic lupus erythematosus in California and Pennsylvania in 2000: estimates obtained using hospitalization data. Arthritis Rheum 2007; 56: 2092–2094.
- [4] Sherer Y, Gorstein A, Fritzler M, Shoenfeld Y. Autoantibody explosion in systemic lupus erythematosus: More than 100 different antibodies found in SLE patients. Sem Arthritis Rheum 2004; 34: 501–507.
- [5] Fritzler MJ, Elkon KB. Autoantibodies in SLE. In Hochberg, MC., et al. Rheumatology. Elsevier, 2008, p. 1253–1264.
- [6] Urowitz MB, Gladman DD, Tom BD, et al. Changing patterns in mortality and disease outcomes for patients with systemic lupus erythematosus. J Rheumatol 2008; 35: 2152–2158.
- [7] Gladman DD, Urowitz MB. Clinical features of systemic lupus erythematosus. In Hochberg, MC., et al. Rheumatology. Elsevier, 2008, p. 1277–1297.
- [8] Lahita R, et al. The Clinical Presentation in SLE in Systemic lupus erythematosus, Fifth edition 2011, Elsevier Inc, 525–570.
- [9] Mulherin D, Bresnihan B. Systemic lupus erythematodes Bailliere´s. Clin Rheumatol 1991; 7: 31–57.
- [10] Gillian JN, Sontheimer RD. Skin manifestations of SLE. Clin Rheum Dis 1981; 8: 207–218.
- [11] Sontheimer RD. The lesions of cutaneous lupus erythematosus – a review and personal perspective on nomenclature and classification of cutaneous manifestations of lupus erythematosus. Lupus 1997; 6: 84–95.
- [12] The American College of Rheumatology nomenclature and case definitions for neuropsychiatric lupus syndromes. Arthritis Rheum 1999; 42: 599–608.
- [13] Bertsias G, Ioannidis JPA, Aringer M, et al. EULAR recommendations for the management of systemic lupus erythematosus with neuropsychiatric manifestations: report of a task force of the EULAR standing committee for clinical affairs. Ann Rheum Dis 2010; 69: 2074–2082.
- [14] Weening JJ, D’Agati VD, Melvin M, et al. The classification of glomerulonephritis in systemic lupus erythematosus revisited. Kidney Int 2004; 65: 521–530.
- [15] Horák P, Tegzová D, Závada J a spol. Doporučení ČRS pro diagnostiku a sledování nemocných se SLE. Čes Revmatol 2013; 2: 59–70.
- [16] Tan EM, Cohen AS, Fries JF, et al. The 1982 revised criteria for the classificatin of systemic lupus erythematosus. Arthritis Rheum 1982; 25: 1271–1277.
- [17] Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1997; 40: 1725–1726.
- [18] Petri M, Orbai AM, Alarcon GS, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum 2012; 64: 2677–2686.
- [19] Gordon C. Long‑term complications of systemic lupus erythematosus. Rheumatology 2002; 41: 1095–1100.
- [20] Ruiz‑Irastorza G, Lima F, Alves J, et al. Increased rate of lupus flare during pregnancy and the puerperium. A prospective study of 78 pregnancies. Br J Rheumatol 1996; 35: 133–139.
- [21] Dostál C, Vencovský J. Těhotenství a SLE. In: Systémový lupus erythematodes. Medprint, 1997: 147–153.
- [22] Skorpen GC, Hoeltzenbein M, Tincani A, et al. The EULAR point to consider for use of antirheumatic drugs before pregnancy and during pregnancy and lactation. Ann Rheum Dis 2016, 0: 1–16.
- [23] Petri M, Howard D, Repke J. Frequency of lupus flare in pregnancy: The Hopkins lupus pregnancy centre experience. Arthritis Rheum 1991; 34: 1358–1364.
- [24] Urowitz MB, Gladman DD, Farewell VT, et al. Lupus and pregnancy studies. Arthritis Rheum 1993, 36: 1392–1396.
- [25] Caritis S, Sibai B, Hauth J, et al. Low aspirin to prevent preeclampsia in women at high risk. N Engl J Med 1998; 338: 701–705.
- [26] Hunt BJ, Doughty H, Majumdar G, et al. Thromboprophylaxis with low molecular weight heparin in high‑risk pregnancy. Thrombo Haemost 1997; 77: 39–43.
- [27] Brester JA, Quenby SM. Alfirevic Z, et al. Intra‑uterine death due to umbilical cord trombosis secondary to antiphospholipid syndrome. Lupus 1999; 8: 558–559.
- [28] Backos M, Rai R, Regan L, et al. Pregnancy complications i women with recurrent miscariiage associated with antiphospholipid antibodies treated with low dose aspirin and heparin. Br J Obstet Gynaecol 1999; 106: 102–107.
- [29] Janssen NM, Genta MS. The effects of immunosuppressive and anti‑inflammatory medications on fertility, pregnancy and lactation. Arch Intern Med 2000; 160: 610–619.
- [30] Koh JH, Ko HS, Kwok SK, et al. Hydroxychloroquine and pregnancy on lupus flares in Korean patients with SLE. Lupus 2015; 24: 210–217.
- [31] Ostensen M, Khamasta M, Lockshin, et al. Anti‑inflammatory and immunosuppresive drugs and reproduction. Arthritis Res Ther 2006; 8: 209.
- [32] Clowswe ME, Magder L, Witter F, et al. Hydroxychloroquine in lupus pregnancy. Arhritis Rheum 2006; 54: 3640–3647.
- [33] Langagegaard V, Pedersen L, Gislum M, et al. Birth outcome in women treated with azathioprine or merkaptopurine during pregnancy: a Danish nationwide cohort study. Aliment Pharmacol Ther 2007; 25: 73–81.
- [34] Silva CA, Hilaro MO, Febronio MV, et al. Pregnancy outcome in juvenile SLE: a Brasilian multicenter cohort study. J Rheumatol 2008; 35: 1414–1418.
- [35] Horák P, Tegzová D, Závada J a spol. Doporučení ČRS pro terapii SLE. Čes Revmatol 2013; 3: 110–122.
- [36] Dennis GJ. Belimumab: a BLyS‑specific inhibitor for the treatment of systemic lupus erythematosus. Clin Pharmacol Ther 2012; 91: 143–149.
- [37] Navarra SV, Guzmán RM, Gallacher AE, et al.; BLISS‑52 Study Group. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo‑controlled, phase 3 trial. Lancet 2011; 377: 721–731.
- [38] Furie R, Petri M, Zamani O, et al.; BLISS‑76 Study Group A phase III, randomized, placebo‑controlled study of belimumab, a monoclonal antibody that inhibits B lymphocyte stimulator, in patients with systemic lupus erythematosus. Arthritis Rheum 2011; 63: 3918–3930.
- [39] Merrill JT, Ginzler EM, Wallace DJ, et al. Long‑term safety profile of belimumab plus standard therapy in patients with systemic lupus erythematosus. Arthritis Rheum 2012; 64: 3364–3373.
- [40] Dörner T, Kaufmann J, Wegener WA, et al. Initial clinical trial of epratuzumab (humanized anti‑CD22 antibody) for immunotherapy of systemic lupus erythematosus. Arthritis Res Ther 2006; 8: R74.
- [41] Wallace DJ, Kalunian K, Petri MA, et al. Efficacy and safety of epratuzumab in patients with moderate/severe active systemic lupus erythematosus: results from EMBLEM, a phase IIb, randomised, double‑blind, placebo‑controlled, multicentre study. Ann Rheum Dis 2014; 73: 183–190.
- [42] Ginzler EM, Wax S, Rajeswaran A, et al. Atacicept in combination with MMF and corticosteroids in lupus nephritis: results of a prematurely terminated trial. Arthritis Res Ther 2012; 14: R33.