Protidestičková léčba – kde jsme a co můžeme očekávat?
Antitrombotická, zejména protidestičková léčba hraje v profylaxi pandemie kardiovaskulárních chorob zásadní úlohu. Dosavadní léčebné strategie byly často zatíženy významnou interindividuální variabilitou účinku, u nezanedbatelného procenta nemocných léčba selhávala či se objevovalo neúnosné riziko krvácení. Z těchto důvodů zaznamenáváme významnou aktivitu ve vývoji nových protidestičkových postupů a inovaci ve skupinách již zavedených. Z léků inhibujících adhezi trombocytů k subendoteliálním strukturám je ve fázi klinického hodnocení řada nových molekul na bázi protilátek proti von Willebrandovu faktoru (vWF) či proti vazným receptorům. Významným vylepšením léčby je nástup nových, účinnějších či bezpečnějších protidestičkových léků inhibujících aktivaci trombocytů. Ze skupiny nových inhibitorů destičkových receptorů P2Y 12 byl schválen ke klinickému užití prasugrel a tikagrelor. Z inhibitorů ADP receptorů zůstal pouze kangrelor, vývoj elinogrelu byl ukončen. Významně pokročil vývoj antagonistů trombinových receptorů typu PAR-1 (vorapaxar, atopaxar ve fázi klinického hodnocení) nebo receptorů serotoninových (sarpogrelat). Rovněž strategie cílená na stabilizaci trombocytů zvýšením nabídky cAMP (např. cilostazolem) nabízí nové možnosti. Konečně též ve skupině léků tlumících vlastní agregaci (inhibitory receptorů IIb/IIIa) se po letech objevuje nová perspektiva – kombinovaná molekula biotaparinuxu s tirofibanem (EP224283) s protidestičkovým i antikoagulačním účinkem, kterou je možno účinně neutralizovat.
V etiopatogenezi aterotrombotických příhod hrají významnou úlohu tři „nestabilní“ faktory: prvým je nestabilní plát s obnaženým vysoce trombogenním kolagenem, dále nestabilní céva s dysfunkčním endotelem s potlačenou schopností kontrolovat aktivovanou hemostázu a konečně nestabilní trombocyt aktivovaný např. v rámci akutní cévní příhody, diabetu či cigaretovým kouřem. Zásah na kterékoli etáži snižuje riziko trombotické cévní příhody. Prvé dva faktory optimálně ovlivníme zásahem do aterogeneze – úpravou dyslipidemie a hypertenze, abstinencí od kouření, kontrolou diabetu apod., v prevenci aterotrombotických příhod však hraje prim protidestičková léčba.
S přibývajícími znalostmi o účincích léků si stále více uvědomujeme úskalí, která farmakoterapii provázejí. Zásah do fyziologických pochodů nebývá beztrestný, miliony let trvající vývoj zpravidla volil optimální strategii životních pochodů. Vychýlení rovnováhy, v našem případě primární hemostázy, dovede sice „zkrotit“ hyperaktivované trombocyty a snížit riziko vzniku trombotické příhody, daní však je zvýšené riziko krvácení. Zachováme-li hlavní zásadu medicíny, kterou pregnantně formuloval Purkyně slovy „…ne sit medica gravior ipso morbo“ neboli „…aby léčba nebyla nebezpečnější nežli nemoc“, musí přínos významně převýšit riziko. Konkrétně předpokládaný pokles aterotrombotických příhod musí významně převládat nad závažnými hemoragickými komplikacemi.
Druhým problémem, který si většinou v praxi neuvědomujeme, je plejáda faktorů, které výslednou terapeutickou odpověď ovlivní. Je to především farmakogenetická výbava ovlivňující dostupnost léku, jeho bioaktivaci, farmakodynamický efekt i eliminaci léčiva. Příkladem je ovlivnění účinku klopidogrelu na úrovni transportních systémů kontrolujících jeho dostupnost či metabolických enzymů aktivujících proléčivo. Kombinace polymorfismů snižujících dosažitelnost klopidogrelu a nedostatečně aktivujících mateřskou látku na aktivní metabolit může vést k selhání účinku. Naopak zvýšená resorpce i bioaktivace může vést ke krvácivým komplikacím. Obdobně do děje vstupují lékové interakce. Příkladem je významné snížení resorpce kyseliny acetylsalicylové (ASA) při současném podání inhibitoru protonové pumpy nebo nemožnost inhibice cyklooxygenázy 1 (COX-1) acetylací při komedikaci ASA s některými antiflogistiky, zejména s ibuprofenem.
Jsme tak postaveni před skutečnost, že variabilita genetické výbavy vede k tomu, že zavedená protidestičková léčba je na jedné straně zatížena rizikem selhání účinku, na straně druhé zvýšeným rizikem krvácení. Není proto divu, že vývoj spolehlivějších, účinnějších a bezpečnějších antitrombotik je nesmírně aktivní. V případě inhibice destičkových receptorů P2Y12 pro ADP se vývoj zdařil a nové, „neproblematické“ ireverzibilní (prasugrel) i reverzibilní (tikagrelor) blokátory ADP receptorů již s výhodou užíváme.
Předložený přehled shrnuje současný stav a ukazuje směr dalšího vývoje. Hlavním motem je – vedle vývoje léků se spolehlivějším účinkem, tj. s menší závislostí účinku na farmakogenetické výbavě – inhibice dalších, specifických mechanismů aktivace sekundární hemostázy. Vyvíjeny jsou nové inhibitory vazných trombocytárních receptorů a bivalentních proteinů zprostředkovávajících adhezi trombocytů k subendoteliálním strukturám (inhibitory adheze) či jejich vzájemnou vazbu (inhibitory vlastní agregace). Obdobně jsou ve fázi klinického hodnocení inhibitory receptorů aktivujících trombocyt (inhibitory aktivace) či blokátory destičkových cytokinů či autakoidů (inhibitory degranulace).
Jaké jsou slabiny současných protidestičkových léků?
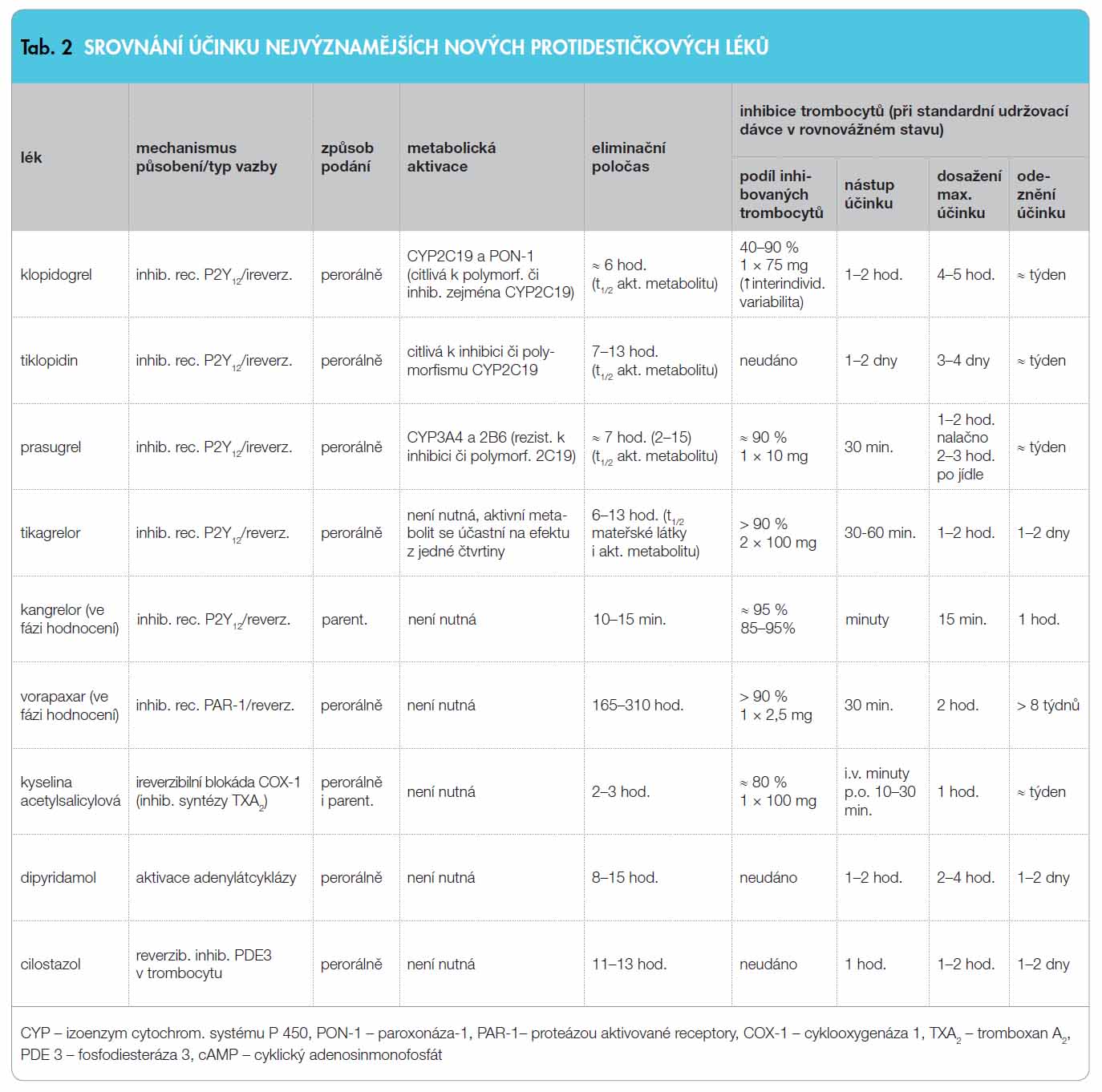
Naše současné spektrum protidestičkových léků (nepřesně označovaných jako antiagregancia) je reprezentováno několika skupinami léků lišícími se mechanismem účinku. Nejdéle užíváme inhibitory aktivace a degranulace trombocytů, a to buď inhibitory aktivace indukované tromboxanem A2 (do této skupiny patří ASA a prakticky neužívané inhibitory tromboxanových receptorů) či inhibitory aktivace zprostředkované adenosindifosfátem (ADP), tedy skupinu blokátorů destičkových receptorů P2Y12 (ireverzibilní – prasugrel a klopidogrel, či reverzibilní – tikagrelor). Inhibitory vlastní agregace jsou reprezentovány blokátory receptorů IIb/IIIa (abciximab, integrilin či tirofiban). Menší význam mají léky stabilizující aktivaci trombocytu zvýšením nabídky cyklického adenosinmonofosfátu (cAMP), jako je dipyridamol a cilostazol, nebo cyklického guanosinmonofosfátu (cGMP), jakými jsou donátory NO (molsidomin).
Protidestičkové léky rutinně užívané k profylaxi aterotrombotických příhod, tj. inhibitory ADP receptorů a ASA, významně sníží výskyt trombotických příhod a mortality – v monoterapii o 20–25 %, v rámci duální léčby o 40–50 %. Nicméně léčba má řadu nedostatků. Nejvýznamnějším a často diskutovaným problémem dvou nejčastěji užívaných protidestičkových léků – ASA a klopidogrelu – je rezistence k léčbě čili nedostatečná odpověď na léčbu, s níž se setkáváme až u třetiny nemocných. Bohužel tato rezistence bývá u řady nemocných na oba léky současně. Proto záměna jednoho léku za druhý či jejich kombinace nemusí problém vyřešit. Podkladem rezistence na ASA může být, vedle nedostatečné spolupráce nemocného, nedostatečná resorpce při vzestupu pH v žaludku, nedostatečná inhibice COX-1 při lékové interferenci nebo zvýšený obrat trombocytů, jako je tomu například u diabetiků. S obdobnými problémy se setkáme u klopidogrelu, zde však hlavní úlohu hraje nedostatečná bioaktivace. Dalším problémem, který je v popředí zejména u klopidogrelu a tiklopidinu, je pomalý nástup účinku. Ve skupině inhibitorů receptorů IIb/IIIa, tedy v pravém smyslu slova antiagregancií, je nevýhodou nutnost parenterálního podání, vyššího krvácivého potenciálu a riziko tzv. rebound fenoménu, tedy protrombotického stavu po jejich vysazení. Společnou Achillovou patou všech dosud užívaných protidestičkových léků je absence antidota. Shrneme-li, pak důvodů pro intenzivní vývoj ve skupině nalezneme dost.
Jaké jsou perspektivy v jednotlivých skupinách protidestičkových léků?
Mechanismem účinku protidestičkových léků je inhibovat primární, tj. destičkovou hemostázu ve fázi adheze, aktivace, degranulace či vlastní agregace trombocytu. Řada postupů je zatím v preklinických fázích, některé již pokročily do klinického hodnocení, jiné jsou již uvolněny ke klinickému užití.
Inhibice adheze trombocytů
Prvá fáze, tj. adheze destiček k sub-endoteliálním strukturám, zejména ke kolagenu, je zprostředkována na jedné straně vazbou multivalentního proteinu von Willebrandova faktoru (vWF) ke komplexu glykoproteinových (GP) receptorů Ib-IX-V, na straně druhé specifickou vaznou doménou ke kolagenu. Paralelně je adheze destičky zprostředkována přímou vazbou trombocytárních GP receptorů VI ke kolagenu (obr. 1a).
Vlastní adhezi umíme ovlivnit na úrovni působení vWF – inhibicí destičkového receptoru Ib v místě vazby na vaznou doménu vWF, blokádou vazné domény vWF či snížením aktivity vWF obsazením většího počtu vazných míst specifickými protilátkami. Vazbu trombocytárního receptoru GP VI pro kolagen se rovněž daří inhibovat monoklonálními protilátkami anti-GP VI.
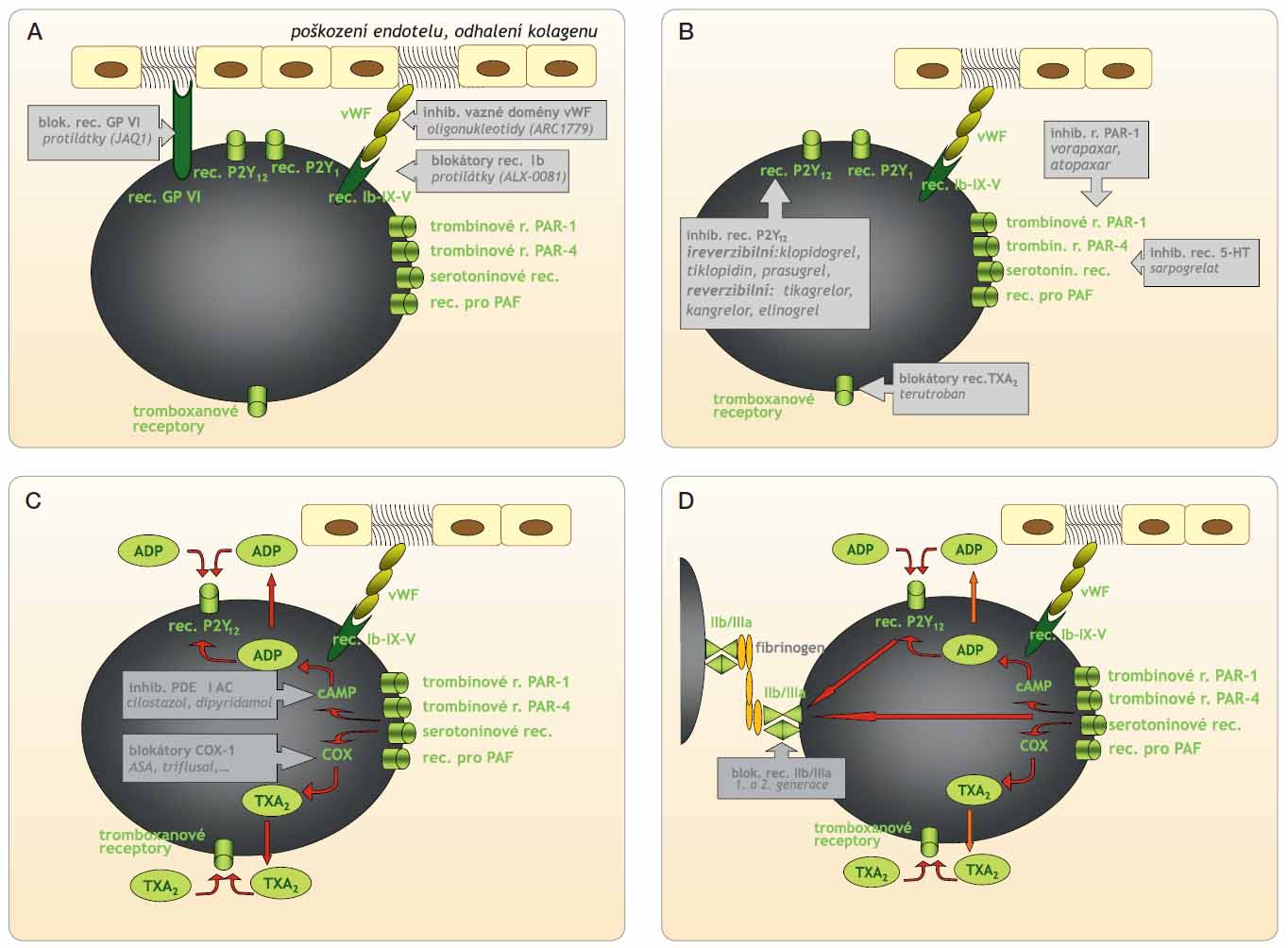 Prvým typem blokátorů adheze jsou inhibitory destičkových receptorů Ib (resp. komplexu GP receptorů Ib-IX-V) typu specifických fragmentů protilátek. Nanoprotilátka ALX-0081 je velmi zajímavou inovací. Jedná se o malý fragment přirozeného řetězce imunoglobulinu, který si ponechal bivalentní afinitu k vaznému místu destičkového GP receptoru Ib s vWF. Obsazením vazby nedojde ani k adhezi, ani k aktivaci trombocytu. Látka ALX-0081 je testována ve II. fázi klinického hodnocení u nemocných s koronárními příhodami typu non-STEMI, kteří jsou léčeni koronární angioplastikou (percutaneous coronary intervention, PCI) [1, 2]. Výsledky zveřejněných testů sledujících efekt in vitro doložily kompletní inhibici adheze i agregace u nemocných po PCI. Ve srovnání se zdravými jedinci však dávky musely být výrazně vyšší [2]. Důvodem byla zřejmě aktivace trombocytů. Druhým směrem testování je útlum zvýšené aktivity vWF při trombofilních mutacích u nemocných s trombotickými mikroangiopatiemi, tj. v profylaxi atak trombotické trombocytopenické purpury. Prověřování účinnosti a bezpečnosti na různých modelech in vivo doložilo lepší vlastnosti, než má většina dosud užívaných protidestičkových léků [3].
Prvým typem blokátorů adheze jsou inhibitory destičkových receptorů Ib (resp. komplexu GP receptorů Ib-IX-V) typu specifických fragmentů protilátek. Nanoprotilátka ALX-0081 je velmi zajímavou inovací. Jedná se o malý fragment přirozeného řetězce imunoglobulinu, který si ponechal bivalentní afinitu k vaznému místu destičkového GP receptoru Ib s vWF. Obsazením vazby nedojde ani k adhezi, ani k aktivaci trombocytu. Látka ALX-0081 je testována ve II. fázi klinického hodnocení u nemocných s koronárními příhodami typu non-STEMI, kteří jsou léčeni koronární angioplastikou (percutaneous coronary intervention, PCI) [1, 2]. Výsledky zveřejněných testů sledujících efekt in vitro doložily kompletní inhibici adheze i agregace u nemocných po PCI. Ve srovnání se zdravými jedinci však dávky musely být výrazně vyšší [2]. Důvodem byla zřejmě aktivace trombocytů. Druhým směrem testování je útlum zvýšené aktivity vWF při trombofilních mutacích u nemocných s trombotickými mikroangiopatiemi, tj. v profylaxi atak trombotické trombocytopenické purpury. Prověřování účinnosti a bezpečnosti na různých modelech in vivo doložilo lepší vlastnosti, než má většina dosud užívaných protidestičkových léků [3].
Druhou možností přístupu je přímá blokáda funkce vWF. Tento vazný protein na jedné straně váže trombocyt k obnaženým kolagenním vláknům, na straně druhé váže další aktivované trombocyty a přispívá k narůstání a ke stabilizaci destičkového trombu. Von Willebrandův faktor můžeme inhibovat monoklonálními protilátkami proti vWF (např. IgG protilátka AJW200) či inhibitory vazných receptorů (např. blokátorem vazné domény vWF k destičkovému receptoru Ib – ARC1779).
Kódovým jménem ARC1779 je označen syntetický oligonukleotid, který je představitelem zmíněných inhibitorů vazné domény vWF. Malé molekuly oligonukleotidů zvané aptamery mají unikátní schopnost inhibovat bioaktivní molekuly, v případě ARC1779 blokují vaznou doménu vWF. Inhibicí kontaktního místa je znemožněna vazba k destičkovému GP receptoru Ib a adheze trombocytu k obnaženému kolagenu. Díky stabilitě, nízké toxicitě a výraznému efektu aptameru na inhibici adheze trombocytů dospěl vývoj do fáze klinického hodnocení. Ve studiích fáze II byla doložena účinnost v prevenci tromboembolie do CNS v průběhu endarterektomie karotidy [4]. Podobně byl doložen účinek na vzestup počtu trombocytů u nemocných s hereditární trombocytopenickou purpurou [5]. Prověřována je účinnost a bezpečnost v indikaci prevence komplikací při intervenci příhod typu non-STEMI či v profylaxi recidivy TIA (tranzitorní ischemické ataky).
Druhou generací inhibitorů vazné domény vWF je sofistikovanější aptamer – ARC15105 – s vyšší inhibiční aktivitou, delší dobou účinku a s dobrou dostupností i při subkutánním podání. Blokáda adheze a agregace byla srovnatelná s abciximabem [6]. Po úspěšném preklinickém testování přechází tento inhibitor vazné domény vWF do fáze klinického hodnocení.
Jak bylo řečeno, druhým způsobem, jak dochází k adhezi trombocytu ke kolagenním vláknům nebo k aktivovaným endoteliím, je přímá vazba trombocytu pomocí receptorů GP VI. Tento mechanismus se uplatňuje zejména při postižení cév aterosklerotickým procesem a může vést k trombotickým komplikacím již v časných fázích aterogeneze, tj. bez přítomnosti ruptury plátu. Testování anti-
trombotického účinku protilátek anti-GP VI pod kódovým označením JAQ1 na modelech aterosklerotického postižení proběhlo úspěšně. Výsledky navíc překvapivě nasvědčují tomu, že vedle inhibice primární hemostázy dochází v modelu na zvířeti také ke zpomalení aterogeneze [7].
Inhibice aktivace destiček
Aktivace trombocytu je kontrolována řadou receptorů (obr. 1b). Aktivace trombocytu je klíčovým krokem, který vede ke změně tvaru (umožňující optimální adhezi ke kolagenu či k jiným povrchům), k aktivaci enzymů (zejm. fosfolipázy C či A2 a adenylátcyklázy) vedoucí k degranulaci a uvolnění řady protrombotických a vazokonstrikčních substancí, k aktivaci povrchových receptorů (konkrétně vazných GP receptorů IIb/IIIa) a ke změně orienta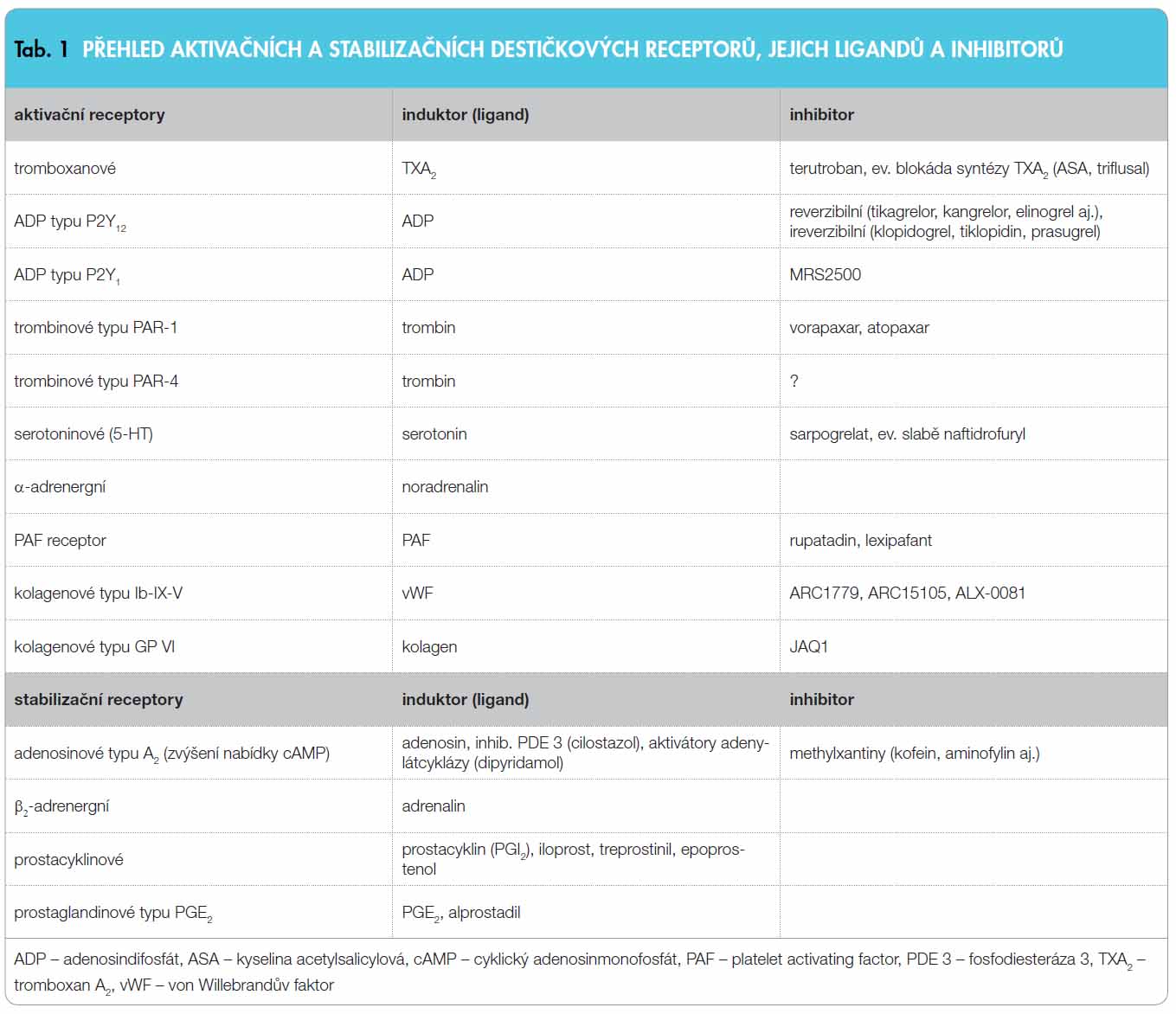 ce membránových fosfolipidů (dovolující vazbu s protrombinázovým komplexem a akcelerující sekundární hemostázu). Nejdůležitějšími aktivačními receptory jsou receptory: tromboxanové, adenosindifosfátové (typu P2Y12 nebo P2Y1), trombinové (typu PAR-1 či PAR-4), serotoninové (5-HT), .-α-drenergní či receptory pro faktor aktivující destičky (PAF). Receptor kolagenový typu Ib-IX-V je nejen aktivační, ale zprostředkovává též adhezi trombocytů k subendoteliálním vrstvám kolagenu. Tyto aktivační receptory jsou za fyziologického stavu v rovnováze se stabilizačními inhibičními receptory: adenosinovými (typ A2A), ß-adrenergními (typu ß2), prostacyklinovými (pro PGI2) či prostaglandinovými (pro PGE2). Jejich přehled představuje tab. 1.
ce membránových fosfolipidů (dovolující vazbu s protrombinázovým komplexem a akcelerující sekundární hemostázu). Nejdůležitějšími aktivačními receptory jsou receptory: tromboxanové, adenosindifosfátové (typu P2Y12 nebo P2Y1), trombinové (typu PAR-1 či PAR-4), serotoninové (5-HT), .-α-drenergní či receptory pro faktor aktivující destičky (PAF). Receptor kolagenový typu Ib-IX-V je nejen aktivační, ale zprostředkovává též adhezi trombocytů k subendoteliálním vrstvám kolagenu. Tyto aktivační receptory jsou za fyziologického stavu v rovnováze se stabilizačními inhibičními receptory: adenosinovými (typ A2A), ß-adrenergními (typu ß2), prostacyklinovými (pro PGI2) či prostaglandinovými (pro PGE2). Jejich přehled představuje tab. 1.
V primární hemostáze jsou významné především ADP receptory typu P2Y12, jejichž stimulace iniciuje dlouhodobou aktivaci trombocytu a expresi agregačních GP receptorů IIb/IIIa. Je proto logické, že nejvíce inovativních molekul působí ve fázi aktivace destičky, především aktivace indukované ADP.
Blokátory receptorů ADP typu P2Y12
Blokáda aktivace trombocytu cestou receptorů ADP patří, vedle blokády tromboxanové cesty, k dnes již klasickým postupům.
Purinergní receptory pro ADP v membráně trombocytu jsou dva – P2Y1 a P2Y12, oba patří mezi receptory spřažené s G-proteinem. První z nich – P2Y1 – otevírá kalciový kanál a zajišťuje rychlý průnik kalcia do cytoplazmy trombocytu s améboidní změnou tvaru a rychlou, ale krátkodobou reverzibilní agregací trombocytu. Druhý receptor – P2Y12 – inhibuje adenylátcyklázu (enzym zvyšující nabídku cAMP a stabilizující trombocyt) a vede k dlouhodobé agregaci. Ke správné funkci primární hemostázy je nutná aktivace obou ADP receptorů, nicméně receptory P2Y12 jsou významnější – stimulují aktivaci trombocytů, potencují degranulaci působků navozenou např. tromboxanem A2 (TXA2) a zejména stabilizují agregáty trombocytů navozené trombinem či TXA2. Inhibice receptoru P2Y1 (např. látkou s kódem MRS2500) se v kontrole hemostázy mnoho neuplatnila, významně však byl snížen průnik leukocytů do sub-endoteliálních prostor.
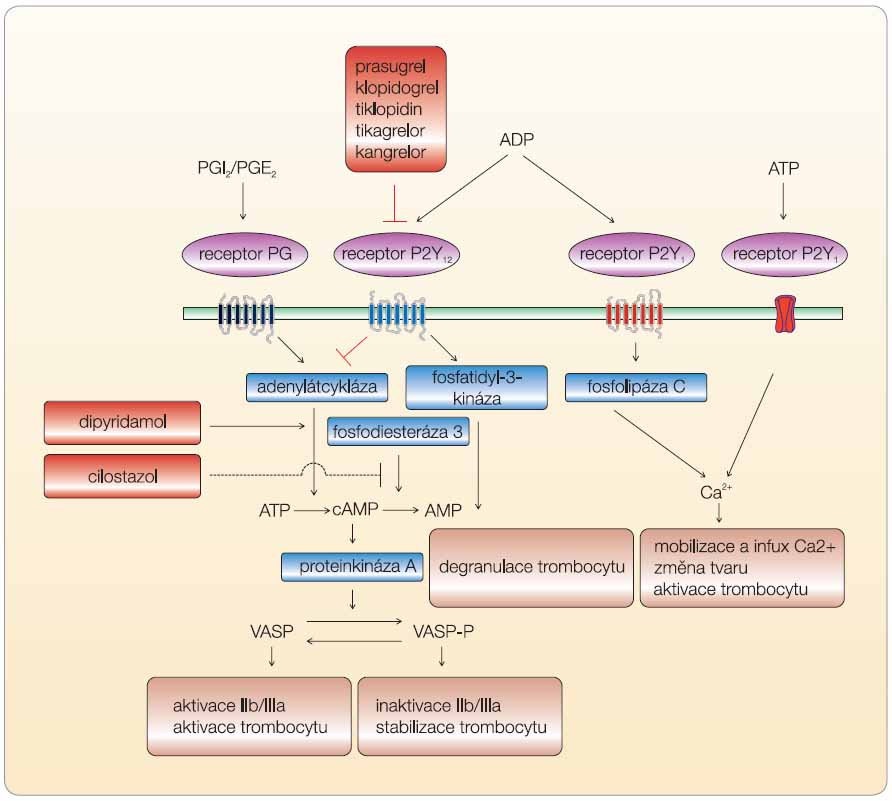 Klinicky významné jsou především ADP receptory typu P2Y12, které jsou aktivovány pomaleji, zato působí dlouhodobě – zprostředkovávají aktivaci a jejich zapojení je klíčovým krokem vlastní agregace. K dosažení rovnováhy jsou protipólem těchto receptorů receptory prostacyklinové. Ty naopak adenylátcyklázu stimulují a trombocyt stabilizují. Posledním poslem, který rozhoduje o aktivaci trombocytu, je regulační protein VASP (vasodilator-stimulated phosphoprotein), poměr fosforylované a defosforylované formy je pro aktivaci určující (obr. 2). Receptory typu P2Y12 můžeme inhibovat ireverzibilně – thienopyridiny, nebo reverzibilně – analogy přirozeného ligandu ADP.
Klinicky významné jsou především ADP receptory typu P2Y12, které jsou aktivovány pomaleji, zato působí dlouhodobě – zprostředkovávají aktivaci a jejich zapojení je klíčovým krokem vlastní agregace. K dosažení rovnováhy jsou protipólem těchto receptorů receptory prostacyklinové. Ty naopak adenylátcyklázu stimulují a trombocyt stabilizují. Posledním poslem, který rozhoduje o aktivaci trombocytu, je regulační protein VASP (vasodilator-stimulated phosphoprotein), poměr fosforylované a defosforylované formy je pro aktivaci určující (obr. 2). Receptory typu P2Y12 můžeme inhibovat ireverzibilně – thienopyridiny, nebo reverzibilně – analogy přirozeného ligandu ADP.
Inhibitory receptorů P2Y12 působí buď nepřímo, tj. po bioaktivaci proléčiva na aktivní metabolit, výsledná blokáda ADP receptoru je nevratná a trombocyt po zbytek cirkulace v krvi neodpovídá na aktivaci ADP. Do této skupiny spadají všechny thienopyridiny (klopidogrel, tiklopidin a prasugrel). Inhibitory přímé – většinou typu purinových analog (tikagrelor či kangrelor) – nepotřebují bioaktivaci, jejich účinek je vratný a doba působení je vázána na dostatečnou plazmatickou hladinu. Prvá skupina má výhodu ireverzibilní vazby na receptor s dlouhodobým protidestičkovým účinkem, daní však je nutnost bioaktivace a pomalejší nástup účinku. Naopak předností přímých a reverzibilních inhibitorů receptorů P2Y12 je rychlý nástup účinku; krátká doba působení však vyžaduje důslednější spolupráci nemocného. Při krvácení nebo nutnosti chirurgického zákroku však může být odeznění efektu během 24 hodin významným přínosem. Antidotum bohužel u stávajících protidestičkových léků nemáme.
Zlatým standardem protidestičkové léčby byla až doposud duální léčba klopidogrelem a ASA. Nicméně, jak bylo řečeno, část nemocných (asi 30 %) nereaguje na klopidogrel optimálně, či neodpovídá na léčbu dostatečně. Při léčbě pak laboratorně prokazujeme vysoké procento aktivovaných trombocytů, klinickým korelátem je menší pokles aterotrombotických příhod ve srovnání s inhibitory ADP receptorů se spolehlivějším efektem. Příčinou nižšího účinku u nezanedbatelné části léčených je nedostatečná nabídka aktivního metabolitu, která by inhibovala funkci asi u 60–70 % trombocytů.
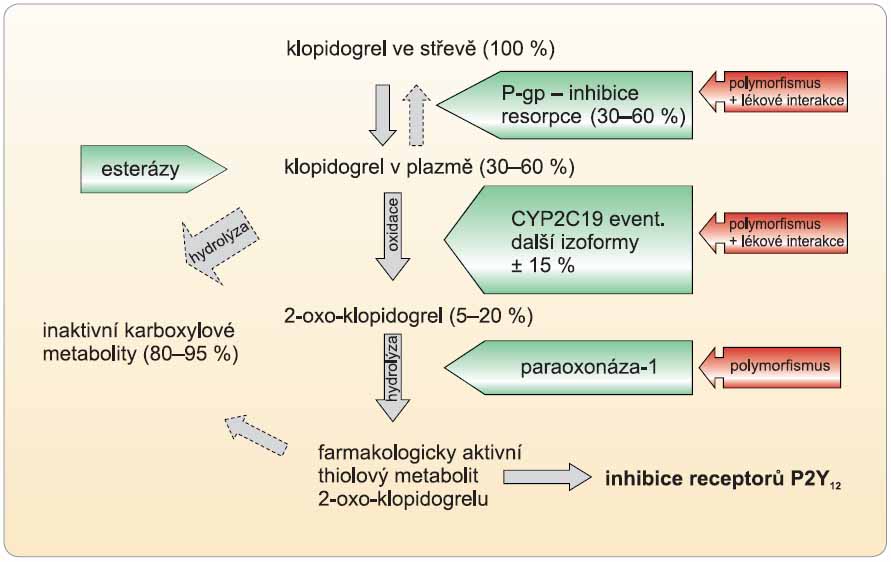 Příčin nedostatečné koncentrace aktivního metabolitu je více. Na úrovni resorpce klopidogrelu působí eliminační pumpa – glykoprotein P (P-gp). Zvýšená aktivita P-gp na podkladě farmakogenetické výbavy sníží dostupnost, naopak inaktivita ji potencuje, rozdíly v dostupnosti jsou více než dvojnásobné. Další a pravděpodobně nejvýznamnější příčinou nedostatečné farmakologické odpovědi však je nedostatečná konverze klopidogrelu na vlastní aktivní metabolit (obr. 3). Za fyziologické situace je asi 85 % proléčiva degradováno esterázami a pouze 15 % je aktivováno na účinný metabolit. V této skutečnosti spočívá kámen úrazu – přeměna na aktivní metabolit neproběhne vždy tak, jak bychom očekávali. Podkladem může být polymorfismus konvertujících enzymů či inhibice konverze některými léky. I zde se uplatňuje řada polymorfismů na úrovni enzymů aktivujících klopidogrel – jak oxidáza CYP 2C19, tak transferáza paroxonáza se vyskytuje ve formách s vyšší, normální a nízkou aktivitou. U nositelů alely CYP2C19*2 nebo CYP2C19*3 (asi u 30 % populace) probíhá konverze velmi pomalu, významně větší část proléčiva je degradována na neúčinné metabolity a efekt klopidogrelu selhává. Vedle genetické výbavy může být na všech úrovních (resorpce i aktivace) problém také s inhibicí či aktivací systémů na podkladě lékové interakce. Nekonzistentní nálezy klinických studií jsou vysvětlitelné sledováním pouze jednoho faktoru, ve hře však je celá mozaika vlivů. Sejde-li se u nemocného léčeného klopidogrelem současně genotyp s nízkou dostupností klopidogrelu (genotyp 3435 T/T P-gp) a současně genotyp s nízkou bioaktivací (alely CYP2C19*2 či CYP2C19*3), setkáme se s nedostatečnou terapeutickou odpovědí. Naopak u genotypu s vysokou dostupností klopidogrelu (genotyp 3435 C/C P-gp) a s vysokou bioaktivací (genotyp CYP2C19*17) je expozice aktivnímu metabolitu klopidogrelu vysoká a setkáme se se zvýšeným rizikem krvácení. Jedna z mála podskupinových analýz ve studii TRITON-TIMI 38, která vzala v úvahu polymorfismus CYP2C19 i P-gp, ukazuje, že kohorta s nízkou expresí P-gp a současně s dobrou funkcí CYP2C19 měla méně než poloviční výskyt závažných vaskulárních příhod (typu MACE) proti podskupině s vysokou expresí P-gp a současně s nízkou aktivitou CYP2C19 – 6,3 % oproti 13,6 % [8].
Příčin nedostatečné koncentrace aktivního metabolitu je více. Na úrovni resorpce klopidogrelu působí eliminační pumpa – glykoprotein P (P-gp). Zvýšená aktivita P-gp na podkladě farmakogenetické výbavy sníží dostupnost, naopak inaktivita ji potencuje, rozdíly v dostupnosti jsou více než dvojnásobné. Další a pravděpodobně nejvýznamnější příčinou nedostatečné farmakologické odpovědi však je nedostatečná konverze klopidogrelu na vlastní aktivní metabolit (obr. 3). Za fyziologické situace je asi 85 % proléčiva degradováno esterázami a pouze 15 % je aktivováno na účinný metabolit. V této skutečnosti spočívá kámen úrazu – přeměna na aktivní metabolit neproběhne vždy tak, jak bychom očekávali. Podkladem může být polymorfismus konvertujících enzymů či inhibice konverze některými léky. I zde se uplatňuje řada polymorfismů na úrovni enzymů aktivujících klopidogrel – jak oxidáza CYP 2C19, tak transferáza paroxonáza se vyskytuje ve formách s vyšší, normální a nízkou aktivitou. U nositelů alely CYP2C19*2 nebo CYP2C19*3 (asi u 30 % populace) probíhá konverze velmi pomalu, významně větší část proléčiva je degradována na neúčinné metabolity a efekt klopidogrelu selhává. Vedle genetické výbavy může být na všech úrovních (resorpce i aktivace) problém také s inhibicí či aktivací systémů na podkladě lékové interakce. Nekonzistentní nálezy klinických studií jsou vysvětlitelné sledováním pouze jednoho faktoru, ve hře však je celá mozaika vlivů. Sejde-li se u nemocného léčeného klopidogrelem současně genotyp s nízkou dostupností klopidogrelu (genotyp 3435 T/T P-gp) a současně genotyp s nízkou bioaktivací (alely CYP2C19*2 či CYP2C19*3), setkáme se s nedostatečnou terapeutickou odpovědí. Naopak u genotypu s vysokou dostupností klopidogrelu (genotyp 3435 C/C P-gp) a s vysokou bioaktivací (genotyp CYP2C19*17) je expozice aktivnímu metabolitu klopidogrelu vysoká a setkáme se se zvýšeným rizikem krvácení. Jedna z mála podskupinových analýz ve studii TRITON-TIMI 38, která vzala v úvahu polymorfismus CYP2C19 i P-gp, ukazuje, že kohorta s nízkou expresí P-gp a současně s dobrou funkcí CYP2C19 měla méně než poloviční výskyt závažných vaskulárních příhod (typu MACE) proti podskupině s vysokou expresí P-gp a současně s nízkou aktivitou CYP2C19 – 6,3 % oproti 13,6 % [8].
Logickým řešením tohoto handicapu klopidogrelu i tiklopidinu je vývoj inhibitorů receptoru P2Y12, jejichž účinek není 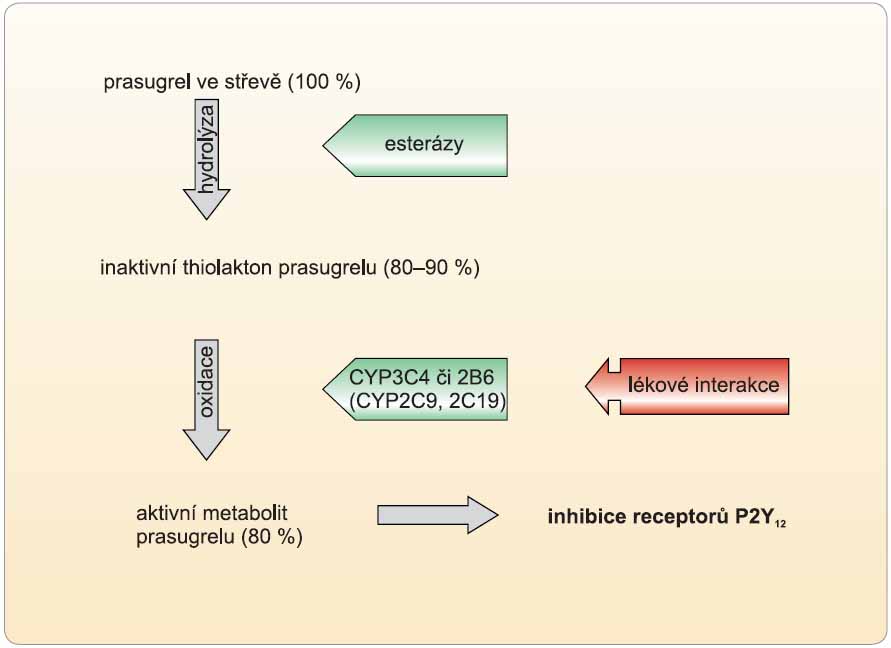 závislý na bioaktivaci. Cesta, jak toho dosáhnout, je dvojí: buď vývojem proléčiva rezistentního k inhibici konverze, nebo zavedením léků, které metabolickou konverzi nevyžadují. Prvou cestu reprezentuje prasugrel – ireverzibilní blokátor dlouhodobě působících receptorů P2Y12, který je představitelem dnes již třetí generace thienopyridinů. Jeho konverze na aktivní metabolit je daleko méně ovlivněna lékovými interakcemi či polymorfismy izoenzymů CYP, efekt je proto spolehlivější a není omezen pouze na populaci s funkčním CYP 2C19 (obr. 4). Při perorálním podání je nástup účinku patrný již za 30 minut a efekt trvá nejméně 72 hodin po ukončení léčby (tab. 2).
závislý na bioaktivaci. Cesta, jak toho dosáhnout, je dvojí: buď vývojem proléčiva rezistentního k inhibici konverze, nebo zavedením léků, které metabolickou konverzi nevyžadují. Prvou cestu reprezentuje prasugrel – ireverzibilní blokátor dlouhodobě působících receptorů P2Y12, který je představitelem dnes již třetí generace thienopyridinů. Jeho konverze na aktivní metabolit je daleko méně ovlivněna lékovými interakcemi či polymorfismy izoenzymů CYP, efekt je proto spolehlivější a není omezen pouze na populaci s funkčním CYP 2C19 (obr. 4). Při perorálním podání je nástup účinku patrný již za 30 minut a efekt trvá nejméně 72 hodin po ukončení léčby (tab. 2).
 Studie TRITON-TIMI 38 prokázala „superioritu“ prasugrelu proti klopidogrelu v léčbě akutních koronárních příhod [9]. Kombinovaný ukazatel efektu (úmrtí, infarkt či iktus) poklesl proti klopidogrelu z 12,1 % na 9,9 % (tj. o absolutní 2,2 %, resp. relativních 19 %). Cenou za tento účinek byl vyšší výskyt významného krvácení o absolutních 0,6 %, respektive relativních 32 %. Tento vzestup odráží skutečnost, že prasugrel je účinný též asi u 30 % nemocných nedostatečně odpovídajících na klopidogrel. Zvýšení rizika krvácení není významné, neboť pravděpodobnost, že zabráníme závažné kardiovaskulární příhodě, je asi 4krát větší ve srovnání s léčbou klopidogrelem nežli riziko, že pacienta poškodíme významným krvácením. Nejvýraznější účinek byl dokumentován u nemocných po implantaci stentu (výskyt trombóz v tepně v oblasti stentu klesl při léčbě prasugrelem ve srovnání s klopidogrelem na polovinu, resp. u stentu lékového poklesl o dvě třetiny). V prasugrelu tak můžeme spatřovat perspektivní lék se spolehlivějším účinkem a rychlejším nástupem efektu – hlavní místo je v léčbě akutních koronárních stavů a lze očekávat doložení účinku také u intervencí v tepenném systému.
Studie TRITON-TIMI 38 prokázala „superioritu“ prasugrelu proti klopidogrelu v léčbě akutních koronárních příhod [9]. Kombinovaný ukazatel efektu (úmrtí, infarkt či iktus) poklesl proti klopidogrelu z 12,1 % na 9,9 % (tj. o absolutní 2,2 %, resp. relativních 19 %). Cenou za tento účinek byl vyšší výskyt významného krvácení o absolutních 0,6 %, respektive relativních 32 %. Tento vzestup odráží skutečnost, že prasugrel je účinný též asi u 30 % nemocných nedostatečně odpovídajících na klopidogrel. Zvýšení rizika krvácení není významné, neboť pravděpodobnost, že zabráníme závažné kardiovaskulární příhodě, je asi 4krát větší ve srovnání s léčbou klopidogrelem nežli riziko, že pacienta poškodíme významným krvácením. Nejvýraznější účinek byl dokumentován u nemocných po implantaci stentu (výskyt trombóz v tepně v oblasti stentu klesl při léčbě prasugrelem ve srovnání s klopidogrelem na polovinu, resp. u stentu lékového poklesl o dvě třetiny). V prasugrelu tak můžeme spatřovat perspektivní lék se spolehlivějším účinkem a rychlejším nástupem efektu – hlavní místo je v léčbě akutních koronárních stavů a lze očekávat doložení účinku také u intervencí v tepenném systému.
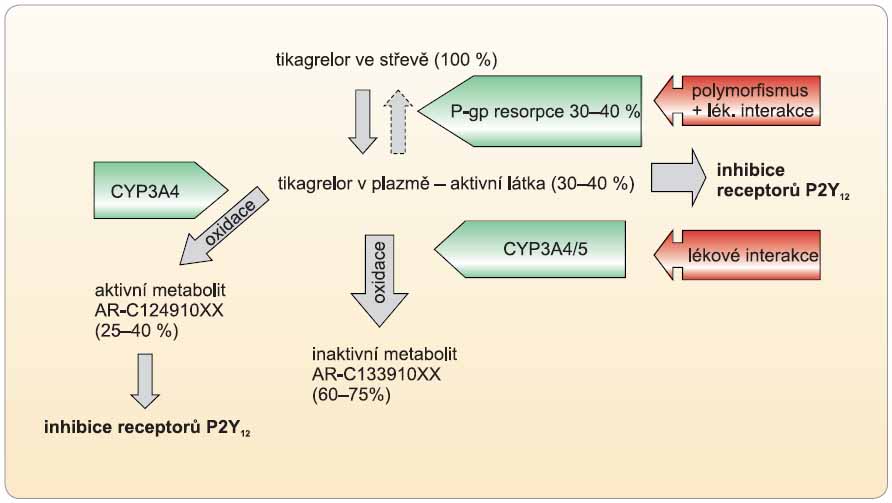 Jiným způsobem vyřešily problémy s bioaktivací non-thienopyridinové inhibitory ADP receptorů P2Y12 tikagrelor či kangrelor, respektive i elinogrel (vývoj ukončen). Hlavními rozdíly od thienopyridinů jsou reverzibilita inhibice a skutečnost, že léky nemají charakter proléčiva, a není proto potřeba konverze na aktivní metabolit. Díky tomu, že nedochází k inhibici konverze, je protidestičkový efekt ve srovnání s klopidogrelem spolehlivější při srovnatelném, či dokonce nižším riziku krvácení (obr. 5).
Jiným způsobem vyřešily problémy s bioaktivací non-thienopyridinové inhibitory ADP receptorů P2Y12 tikagrelor či kangrelor, respektive i elinogrel (vývoj ukončen). Hlavními rozdíly od thienopyridinů jsou reverzibilita inhibice a skutečnost, že léky nemají charakter proléčiva, a není proto potřeba konverze na aktivní metabolit. Díky tomu, že nedochází k inhibici konverze, je protidestičkový efekt ve srovnání s klopidogrelem spolehlivější při srovnatelném, či dokonce nižším riziku krvácení (obr. 5).
Do klinického užití již byl zaveden tikagrelor v indikaci prevence trombotických příhod u nemocných s akutními koronárními příhodami typu nestabilní anginy pectoris a infarktu myokardu bez elevace úseku ST či s elevací úseku ST, kteří jsou indikováni k intervenční nebo konzervativní léčbě. Nástup účinku tikagreloru je po perorálním podání možno pozorovat do 30 minut až 1 hodiny, maximální účinek pak za 1–2 hodiny. Vzhledem k reverzibilitě vazby na receptor je délka efektu kratší a středně dlouhý eliminační poločas (t1/2 6–13 hodin) vede k podávání dvakrát denně (tab. 2).
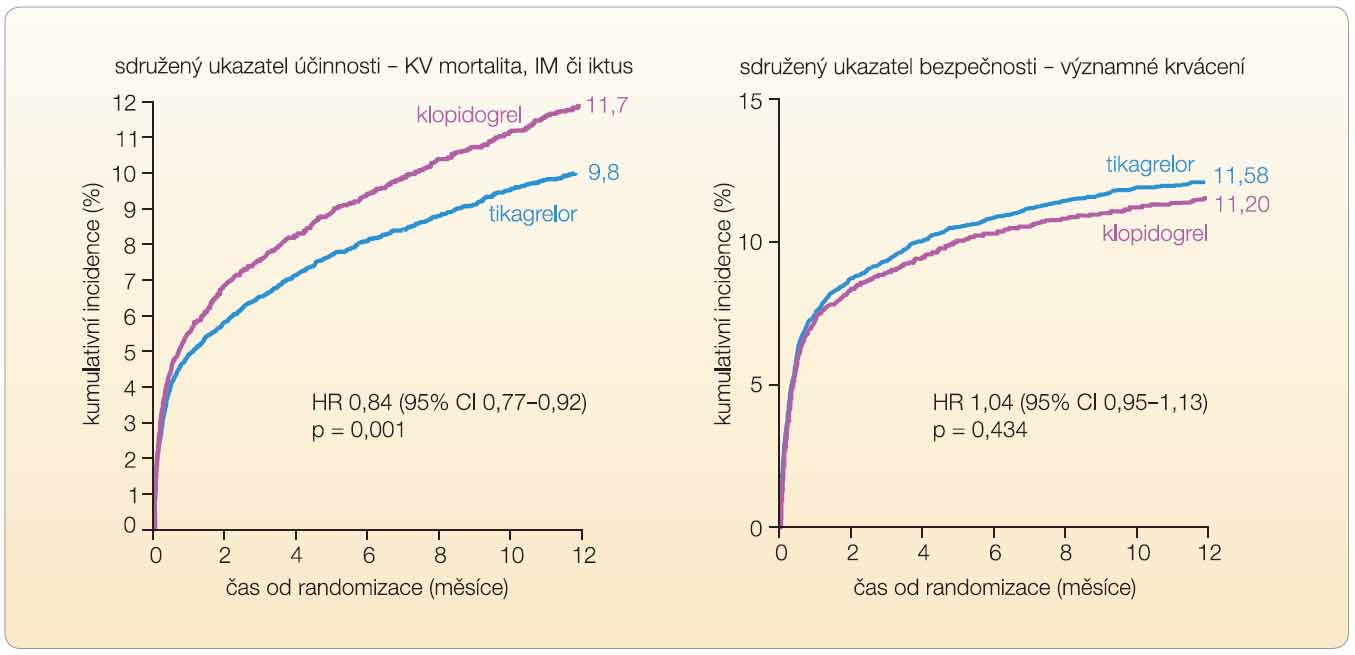 V megastudii PLATO byl prověřován efekt u akutních koronárních příhod v obdobném schématu jako ve studii TRITON-TIMI 38, tedy ve srovnání s léčbou klopidogrelem při bazální léčbě ASA [10]. Kombinovaný ukazatel efektu – KV mortalita, výskyt infarktu myokardu či iktu – poklesl významně o relativních 16 % či o absolutních 1,9 % (graf 1a). Výskyt krvácení byl přitom v obou větvích srovnatelný (graf 1b). V porovnání s klopidogrelem prokázal tikagrelor při srovnatelné bezpečnosti vyšší účinnost. Ve srovnání s prasugrelem byl dopad na pokles závažných vaskulárních příhod obdobný, výskyt závažných krvácivých příhod však byl nižší.
V megastudii PLATO byl prověřován efekt u akutních koronárních příhod v obdobném schématu jako ve studii TRITON-TIMI 38, tedy ve srovnání s léčbou klopidogrelem při bazální léčbě ASA [10]. Kombinovaný ukazatel efektu – KV mortalita, výskyt infarktu myokardu či iktu – poklesl významně o relativních 16 % či o absolutních 1,9 % (graf 1a). Výskyt krvácení byl přitom v obou větvích srovnatelný (graf 1b). V porovnání s klopidogrelem prokázal tikagrelor při srovnatelné bezpečnosti vyšší účinnost. Ve srovnání s prasugrelem byl dopad na pokles závažných vaskulárních příhod obdobný, výskyt závažných krvácivých příhod však byl nižší.
Zajímavé je pozorování zvýšeného výskytu asymptomatických AV blokád a dušnosti, které se objevují v prvé periodě po zahájení léčby tikagrelorem. Vysvětlením by mohla být spekulace vyslovená na základě experimentálních prací, že tikagrelor není pouze inhibitorem receptoru P2Y12, ale stimuluje uvolnění adenosinu, resp. ATP z erytrocytů [33]. Stimulace adenosinových receptorů může vést k aktivaci dechového centra, ke zvýšení bronchospastické aktivity vedoucí k bronchokonstrikci či k navození bradykardie až poruchy síňo-komorového vedení.
Dalším reverzibilním non-thienopyridinovým inhibitorem ADP receptoru P2Y12 je kangrelor – obdobně jako tikagrelor nevyžaduje bioaktivaci. Jeho předností je velmi rychlý nástup účinku (v řádu minut), účinek je však krátký, odezní během desítek minut (tab. 2). Vzhledem ke krátkému efektu a nutnosti kontinuální parenterální aplikace byl testován u akutních koronárních příhod (studie CHAMPION PLATFORM) jen v akutním, periprocedurálním období. V jedné větvi byl u nemocných s akutní koronární příhodou indikovaných k provedení PCI aplikován kangrelor před výkonem (bolus + infuze) s následnou léčbou klopidogrelem, ve druhé byli pacienti léčeni jen klopidogrelem. Při sledování celkové mortality, infarktu myokardu či nutnosti revaskularizace se v prvých 48 hodinách nepotvrdil předpoklad příznivějšího působení kangreloru na pokles mortality a morbidity, krvácivých příhod přitom významně přibylo [11]. Po zklamání u akutních koronárních stavů se další vývoj klinického hodnocení orientuje na situace, kdy je potřeba krátkodobého efektu, například v době před revaskularizační operací.
Posledním přímým reverzibilním inhibitorem receptorů P2Y12 byl elinogrel. V srpnu 2011 bylo avizováno pozastavení studií 3. fáze klinického hodnocení a v lednu 2012 bylo oznámeno ukončení vývoje.
Jak vzájemně porovnat dostupné inhibitory ADP receptorů P2Y12? Hlavní předností ireverzibilních blokátorů – prasugrelu, klopidogrelu a tiklopidinu – je zajištění blokády trombocytů i při prodloužení dávkovacího intervalu či při vynechání dávky. Inhibice trombocytů trvá ještě několik dnů po vysazení dávky. Tato vlastnost však, při neznalosti antidota, může být na závadu v případě potřeby rychlého ukončení účinku, například při krvácení, traumatu či při potřebě náhlé chirurgické intervence. Druhou nevýhodou je nutnost metabolické konverze na vlastní aktivní látku. Právě zde je hlavní slabina klopidogrelu – při bioaktivaci je potřeba dvou metabolických kroků, kde jsou zapojeny enzymy s výraznou polymorfií. Podobně je farmakogenetickými vlivy (na úrovni P-gp) ovlivněna biologická dostupnost klopidogrelu.
Prasugrel, který podáváme též jako proléčivo, nemá metabolickou konverzi závislou na polymorfních enzymech a jeho biologická dosažitelnost není snižována P-gp. Jeho účinek je tak spolehlivý. Efekt na celé spektrum populace, nehledě na genotyp, je hlavní předností prasugrelu. Je-li dosaženo účinku u dalších 30 % nemocných, kteří neodpovídali na léčbu klopidogrelem a kteří odpovídají na prasugrel, musíme počítat s vyšším výskytem krvácení – skutečně krvácivé komplikace jsou asi o třetinu častější. Lze tedy říci, že krvácivý potenciál klopidogrelu a prasugrelu u osob, kde je lék účinný, je plně srovnatelný.
Jediným představitelem reverzibilních inhibitorů uvolněným k užití v klinické medicíně je a pravděpodobně po řadu let zůstane tikagrelor. Jeho výhodou je kratší doba účinku, omezená při běžném dávkování na 24 hodin, rychlejší nástup účinku a absence potřeby biokonverze na aktivní metabolit. Reverzibilita blokády receptoru může být výhodou – potřebujeme-li ukončit inhibici primární hemostázy, např. při akutní indikaci chirurgického zákroku, zejména revaskularizace myokardu. Na druhé straně efekt závisí na dokonalé spolupráci nemocného, vynechání více než jedné dávky vede k obnovení aktivity trombocytů a k riziku trombotické komplikace. Odpadá riziko vyplývající z rezistence kvůli polymorfní biokonverzi či navozené lékovými interakcemi.
Srovnáme-li hlavní představitele inhibitorů ADP receptorů – klopidogrel, prasugrel a tikagrelor – z hlediska hlavní indikace, jíž je profylaxe trombotické tepenné okluze po akutních koronárních příhodách, můžeme konstatovat větší efekt na pokles výskytu kardiovaskulárních příhod a mortality u tikagreloru a prasugrelu oproti klopidogrelu. Skutečně v situaci, kde je výskyt rozhodující měrou ovlivněn primární hemostázou, jako je tomu např. při trombóze v místě implantace koronárního stentu, pozorujeme pokles počtu trombotických příhod o třetinu až polovinu. V jiných indikacích, kde je nutno počítat s multifaktoriální závislostí (ovlivnění mortality, infarktu či iktu), se rozdíl pohybuje mezi 15–20 %. Společná data pro tikagrelor a prasugrel dokládají pokles celkové mortality, infarktu či iktu o 17 %, celkové mortality také o 17 %, nefatálního infarktu myokardu o 21 % či trombózy ve stentu o 39 % ve srovnání s dosud „zlatým standardem“ – klopidogrelem. Při nepřímém porovnání tikagreloru, resp. prasugrelu vůči klopidogrelu nebyl prokázán rozdíl mezi tikagrelorem či prasugrelem v rámci hodnocení celkové mortality, výskytu nefatálního infarktu myokardu či nefatálního iktu. Rozdíl byl pozorován pouze v nižším výskytu trombotických příhod v místě implantovaného stentu (ve prospěch prasugrelu) a celkového výskytu krvácení (ve prospěch tikagreloru). Redukce krvácivých komplikací byla dána zejména poklesem počtu příhod spojených s kardiochirurgickými výkony, krvácení mimo tyto výkony bylo u obou léků srovnatelné. Také tolerance obou léků byla srovnatelná, procento nemocných, kteří přerušili léčbu, bylo identické. Lze konstatovat, že tikagrelor má ve srovnání s prasugrelem přednost nižšího výskytu krvácivých komplikací spojených zejména s kardiochirurgickými zákroky, prasugrel má naopak výhodu nižšího výskytu trombóz v místě implantovaného stentu. Účinnost a bezpečnost v ostatních indikacích je srovnatelná.
Blokátory trombinových receptorů PAR-1
Aktivace trombocytu vytvořeným trombinem propojuje sekundární hemostázu s primární. V současné době jsou ve fázi klinického hodnocení dva přípravky inhibující trombinový receptor PAR-1 (protease-activated receptor 1) obr. 1b. Vzhledem k tomu, že trombinové receptory nemají vliv na adhezi trombocytů, je pravděpodobné, že místo inhibitorů receptoru PAR-1 zůstane v kombinaci s blokátory ADP či s blokátory tromboxanové cesty aktivace. Ve třetí fázi klinického hodnocení je účinná látka vorapaxar a 2. fáze hodnocení je ukončena u atopaxaru.
Vorapaxar, selektivní reverzibilní blokátor destičkového receptoru PAR-1, je prověřován v indikaci proxylaxe cévních příhod u aterotrombotických onemocnění. V pokročilejších fázích hodnocení je testován v kombinaci s ostatními protidestičkovými léky, zejména s ASA či klopidogrelem. Tento antagonista destičkového trombinového receptoru má rychlý nástup účinku (do hodiny) a dlouhou dobu působení (t1/2 160–310 hodin) umožňující perorální podávání jedenkrát denně (po aplikaci nasycovací dávky). I když je blokáda receptoru reverzibilní, je zajištěn efekt po dobu několika týdnů (tab. 2). Tato skutečnost může být kritická při současné nedostupnosti antidota.
Ze studií 2. fáze hodnocení je nejvýznamnější TRA-PCI, v níž se ukázala bezpečnost vorapaxaru při předpokladu účinnosti u nemocných s primární PCI. Při úvodní dávce 40 mg a udržovací dávce 2,5 mg se neobjevilo závažné krvácení, výskyt MACE sice ve srovnání s placebem klesl, nicméně čísla byla příliš malá k jakýmkoli závěrům [9, 10]. Podobně klesl výskyt periprocedurálních infarktů v rámci plánované PCI při přidání vorapaxaru ke standardní, zpravidla duální protidestičkové léčbě (16,9 % vs. 42,9 %; p = 0,013) [12]. Na základě příznivých výsledků v indikaci prevence recidivy iktu a infarktu myokardu (při léčbě ASA) či v ovlivnění periprocedurálních infarktů (na pozadí duální protidestičkové léčby) pokročily studie do 3. fáze hodnocení.
Ve studii TRACER byl prověřován efekt vorapaxaru po akutní koronární příhodě typu non-STEMI v kombinaci se standardní, zpravidla duální protidestičkovou léčbou [13]. Studie byla předčasně ukončena z bezpečnostních důvodů – nárůst středně významného a těžkého krvácení činil 35 % (absolutní vzestup o 2 %), intrakraniální krvácení stouplo několikanásobně, v absolutních číslech o 0,9 %. Kombinovaný ukazatel bezpečnosti – KV mortalita, infarkt myokardu či iktus – klesl o 11 % (absolutní pokles o 1,7 %). Při porovnání rizika významného krvácení s přínosem, který představuje pokles počtu trombotických příhod, nebyl zaznamenán jakýkoli prospěch.
Obdobně, ve studii TRA-2oP-TIMI 50 u chronických kardiovaskulárních onemocnění, tj. po překonaném infarktu myokardu, po iktu či s ischemickou chorobou dolních končetin (ICHDK), byl u téměř 27 tisíc nemocných hodnocen pokles řady mortalitně/morbiditních parametrů při stávající protidestičkové léčbě (> 50 % pacientů s duální léčbou) s přidáním vorapaxaru [14]. Výsledky studie byly recentně zveřejněny. Doplnění lege artis léčby v rámci sekundární prevence o vorapaxar vedlo během tří let léčby k významnému poklesu kombinovaného ukazatele účinnosti (KV mortalita, infarkt myokardu a mozková příhoda) o 13 % (RR 0,87, 0,80–0,94; p < 0,001), absolutní pokles byl o 1,2 % (9,3 % vs. 10,5 %). Bohužel zejména ve skupině nemocných s předchozí mozkovou příhodou a u nemocných s nízkou hmotností (< 60 kg) stoupl neúnosně (0,9 % vs. 2,4 %) výskyt hemoragických iktů. Pokud se tato podskupina vyloučila, pak na každých 1000 léčených po dobu 2–3 let bylo zabráněno 19 významným kardiovaskulárním příhodám (KV mortalita, infarkt myokardu, iktus) za cenu 2 hemoragických mozkových příhod a 10 středně významných až těžkých krvácivých příhod. Při sledování součtu závažných krvácivých příhod a závažných kardiovaskulárních příhod byl v této podskupině zaznamenán významný pokles o 16 %. Při sledování optimální subpopulace s příznivým poměrem efekt/riziko se ukázal příznivý poměr zejména u nemocných indikovaných pro překonaný infarkt myokardu, bez anamnézy iktu, s hmotností nad 60 kg a ve věkové kategorii pod 75 let.
Druhým inhibitorem receptorů PAR-1 je atopaxar, který vedle blokády aktivace destičky tlumí též uvolnění molekul akutní fáze, zejména cytokinů interleukinu-6, sCD40L a P-selektinu. Klinické hodnocení 2. fáze proběhlo ve třech větvích programu LANCELOT. U nemocných s akutními koronárními příhodami (studie LANCELOT-ACS) byl pozorován nižší výskyt ischemických epizod a trend k poklesu počtu příhod, výskyt krvácivých komplikací se nezvýšil [15]. Ve druhé větvi programu (LANCELOT-CAD) byla opět hledána optimální dávka atopaxaru, tentokrát u stabilizovaných forem ICHS. Přidání atopaxaru ke standardní léčbě vedlo k nesignifikantnímu poklesu počtu trombotických příhod a k významnému vzestupu výskytu méně významných epizod krvácení, počet závažných krvácení se nezvýšil [16]. Třetí větev byla věnována opět nemocným s akutními koronárními příhodami, tentokrát u japonské populace (J-LANCELOT). Studie 3. fáze hodnocení zatím nejsou v centrálním registru (clinicaltrials.gov) avizovány.
Destičkové receptory PAR-4 mají zřejmě jen podpůrný účinek. Po inhibici receptoru PAR-1 sice může být trombocyt aktivován stimulací receptorů PAR-4, nicméně dávka trombinu musí být mnohonásobně vyšší. O možnostech farmakologické blokády těchto receptorů nejsou publikovány žádné zprávy.
Blokátory tromboxanových receptorů a inhibitory syntézy TXA2
Tlumit primární hemostázu na úrovni tromboxanové cesty aktivace můžeme dvojím způsobem. Prvým je inhibice syntézy tromboxanu A2 (TXA2), druhou je blokáda jeho receptorů na úrovni trombocytu či cévní stěny (obr. 1b a 1c).
Blokáda COX-1 kyselinou acetylsalicylovou patří k nejstarší strategii protidestičkového působení. Podkladem účinku je acetylace serinu v oblasti katalytického místa COX-1, enzymu umožňujícího konverzi kyseliny arachidonové na prostaglandin H2. Ten je prekurzorem jak vazodilatačně, protektivně a antiagregačně působících prostaglandinů PGI2 (prostacyklinu) a PGE2, tak i jejich protějšku, proagregačně a vazokonstrikčně působícího tromboxanu A2 (TXA2). Blokáda COX-1 tak má dvojí ostří – na jedné straně pozitivně a dlouhodobě ireverzibilně ovlivňuje aktivaci trombocytů, na druhé straně – do resyntézy nové COX-1 v endoteliích může zhoršit funkci endotelu. Vedle ne zcela ideálního farmakologického působení ASA se setkáváme také s problémy s dostupností ASA. Slabé kyseliny hydrofilní povahy, jakou je ASA, jsou vstřebávány pouze v nedisociovaném stavu, tj. při pH nižším než 3,5, tedy resorpce se uskuteční optimálně ve velmi kyselém prostředí žaludku. Při žaludečním pH převyšujícím 3,5, jak je běžné při podávání inhibitorů protonové pumpy (IPP), je ASA disociována a vstřebává se výrazně hůře. Klinický význam snížení dostupnosti ASA při současném podávání IPP dokládá zajímavá retrospektivní analýza registru 100 tisíc dánských nemocných po infarktu myokardu. Z dvaceti tisíc nemocných, kteří byli léčeni ASA (zpravidla 75 mg denně) bez aplikace inhibitorů ADP receptorů, bylo 22 % léčeno také IPP. Ve skupině léčené kombinací ASA + IPP byla pozorována vyšší incidence kombinovaného ukazatele, jímž je kardiovaskulární mortalita, infarkt myokardu a iktus, o 46 %, resp. při přesnější „case-matched“ analýze vzrostl výskyt dokonce o 61 %. Zvýšení mortality/morbidity šlo napříč skupinou IPP, nezávisle na podaném přípravku [17]. Z těchto důvodů jsou hledány nové cesty inhibice působení tromboxanu.
Novinek ve skupině inhibitorů syntézy TXA2 není mnoho. Alternativou ASA je triflusal. Tento derivát ASA rovněž preferenčně a ireverzibilně inhibuje COX-1. Inhibice COX-1 je sice slabší než po podání ASA, nicméně tento nedostatek bohatě kompenzuje skutečnost, že se jedná o blokádu semiselektivní. To znamená, že je potlačena syntéza trombocytární cyklooxygenázy, tedy syntéza TXA2, ale v endoteliích je COX-1 tlumena o řád méně, a blokáda syntézy prostacyklinu je proto nevýznamná. Navíc triflusal stimuluje NO-syntázu a zvýšenou nabídkou oxidu dusnatého funkci endotelu zlepšuje.
Potenciálem triflusalu by mohla být protidestičková léčba nemocných s intolerancí ASA. Jako léčebnou alternativu jej poslední doporučení neuvádějí [18]. Triflusal byl testován zejména v prevenci recidivy mozkových příhod v několika studiích (TACIP, TAPIRSS), doložen byl pokles výskytu mozkových krvácení, nebyl však ve srovnání s ASA pozorován pokles výskytu recidivy mozkových příhod, výskyt infarktu myokardu či pokles mortality [19].
Vedle inhibice syntézy TXA2 inhibicí COX je možno blokovat i tromboxanové receptory. Nadějným antagonistou těchto receptorů byl terutroban. Tento selektivní antagonista TP receptorů (tj. receptorů pro vazokostrikční a proagregační prostanoidy TXA2 a PGG2 či PGH2) má v experimentu antitrombotické a vazodilatační vlastnosti. Klinický účinek byl prověřován proti standardní dávce ASA ve studii PERFORM u téměř 20 tisíc nemocných po ischemické mozkové příhodě [20]. Studie byla předčasně po více než dvou letech ukončena pro shodný účinek na pokles výskytu mozkových příhod, infarktu myokardu či úmrtí z cévních příčin v obou větvích. Výskyt krvácení mírně stoupl v terutrobanové větvi. Zprávy o pokračování v klinickém hodnocení terutrobanu nejsou, vývoj je pravděpodobně zastaven.
Kombinaci blokády syntézy tromboxanu (inhibicí tromboxan-syntázy) a blokády TP receptorů (TXA2/PGH2-receptorů) navodí staronový lék pikotamid. Staronový proto, že po dvou desetiletích stagnace je v rámci profylaxe trombotických příhod u nemocných s ICHDK zmíněn jako protidestičková alternativa v posledních, tj. 9. doporučeních ACCP (American College of Chest Physicians) [21]. Doklady o efektu vycházejí ze studie DAVID, kde byl pikotamid užit u diabetiků s ICHDK. Ve srovnání s ASA byl pozorován pokles celkové mortality o 45 %, výskyt KV příhod však významně ovlivněn nebyl [22]. Vzhledem k tomu, že studie nebyla velká (jen 1200 nemocných), nelze pokládat výsledek za plně validní a uvedení v doporučeních je spíše překvapivé.
Blokátory serotoninových receptorů
Aktivace trombocytů serotoninovými receptory se pokládá za méně významnou (obr. 1b). Blokáda serotoninových receptorů ketanserinem či naftidrofurylem ovlivňuje aktivaci trombocytů jen okrajově, pozorováno bylo jen mírné zpomalení rychlosti agregace. Zajímavou a slibnou inovací by však mohl být sarpogrelat, selektivní antagonista destičkových a kardiálních serotoninových receptorů 2A. Zdá se, že účinek sarpogrelatu může být širší, pozorován byl účinek na prevenci koronárních spasmů a protekce postperfuzního poškození myokardu [23]. Příznivé výsledky studií 2. fáze klinického hodnocení umožnily přechod do fáze předregistračních studií, konkrétně je ohlášena studie u 8 tisíc nemocných s ICHDK [24]. Již dokončená studie u 1500 nemocných léčených v rámci sekundární prevence mozkových příhod (S-ACCESS) však nedoložila výhodu sarpogrelatu proti ASA, výskyt cerebrálních infarktů byl nevýznamně (o čtvrtinu) zvýšen, vaskulární příhody byly v obou skupinách stejně časté, významně (asi o třetinu) se však snížil výskyt krvácení [25]. Pokud se efekt sarpogrelatu potvrdí, pak by blokáda serotoninových receptorů 2A doplnila strategii blokády aktivace trombocytu o další možnost. Trochu překvapivá je skutečnost, že zatím je v klinických hodnoceních prověřován účinek proti ASA, ale neprobíhají žádné studie, kde by byl sarpogrelat podáván spolu s ASA. Přitom studie 2. fáze ukázaly velmi dobrou potenciaci účinku, například v testech in vitro u nemocných s anginou pectoris byl doložen významný pokles počtu destičkových agregátů a potenciace fibrinolytické aktivity endotelu [26].
Blokátory receptorů pro PAF
Inhibice působení PAF (platelet-activating factor) v indikaci protidestičkové léčby zatím mnoho nepokročila. Vzhledem k řadě fyziologických funkcí PAF je efekt jeho inhibice prověřován převážně v jiných indikacích. Blokátory receptorů pro PAF – např. rupatadin (kombinovaný inhibitor histaminových receptorů H1 a receptorů pro PAF) či lexipafant (čistý inhibitor receptorů pro PAF) – mají určitý efekt v léčbě alergických onemocnění či akutní pankreatitidy, avšak antiagregační účinek je zanedbatelný.
Inhibice degranulace trombocytů
Degranulace trombocytů je důležitou složkou primární hemostázy. Byly identifikovány téměř dvě stovky látek, které jsou uvolněny destičkami po aktivaci. Řada z těchto působků má zásadní význam a funguje zpravidla více mechanismy účinku. V prvé řadě aktivují další trombocyty a atrahují je do místa léze. Další důležitou funkcí je vazokonstrikce, neboť omezení průtoku zvýší lokální koncentraci hemostatických působků a proteáz koagulační kaskády, dále lokální vazokonstrikce sníží ztráty krve. Třetí oblastí, kde tyto látky působí, je zánětlivý a reparační proces. Při degranulaci je uvolněn zejména TXA2, ADP či serotonin, tři klíčové molekuly atrahující a aktivující další trombocyty. Paralelní sekrece řady molekul akutní fáze, růstových faktorů nebo vazoadhezivních proteinů se účastní spíše v rámci reparativních pochodů.
Inhibice sekrece je proto důležitou součástí protidestičkové strategie. Hlavní léčebnou strategií není však inhibice syntézy jednotlivých faktorů, ale zabránění aktivaci trombocytu a tlumení děje, který degranulaci iniciuje (obr. 1c). Spíše výjimkou je možnost selektivně ovlivnit jeden působek (zejm. tromboxanu A2) inhibitory COX-1 nebo tromboxan-syntázy (např. kyselinou acetylsalicylovou). Tato léčebná strategie byla probrána výše.
Druhou strategií je stabilizace trombocytu zvýšením nabídky cyklického adenosinmonofosfátu (cAMP) aktivací adenylátcyklázy či cyklického guanosinmonofosfátu (cGMP). Zejména zvýšení koncentrace cAMP v trombocytu má klinický význam, dochází k nižšímu uvolňování adenosindifosfátu a k útlumu aktivace dalších trombocytů.
Prakticky schůdné jsou dva přístupy – zvýšení syntézy cAMP stimulací adenylátcyklázy, která zvyšuje syntézu cAMP, či blokáda degradace cAMP inhibicí fosfodiesterázy (PDE). Na prvé úrovni působí dipyridamol, na druhé cilostazol. Nutno však zdůraznit, že tento postup má spíše podpůrný účinek, základem protidestičkové strategie zůstává působení na mediátory aktivace destiček. Zvýšení nabídky cGMP dosáhneme donátory NO, konkrétně molsidominem. Klinický dopad tohoto postupu nebyl testován, v experimentu má inhibice destiček jen hraniční významnost.
Dostupnou a v neurologické praxi užívanou je kombinace dipyridamolu a ASA. Dipyridamol inhibuje aktivitu enzymu adenosindeaminázy, tímto mechanismem zvyšuje nabídku adenosinu, ten stimuluje adenylátcyklázu, která zvyšuje syntézu cAMP. Právě zvýšení cAMP stabilizuje trombocyt. Dříve předpokládaný efekt dipyridamolu na inhibici PDE, která degraduje cAMP, se nepotvrdil.
Při analýze účinku kombinace ASA a dipyridamolu na cévní komplikace byl doložen pokles výskytu všech závažných cerebrovaskulárních i kardiovaskulárních příhod o 12 %. Při analýze typů příhod se ukázalo, že jediným významným byl snížený výskyt iktů a tranzitorních atak [27]. Metaanalýza doložila významný pokles cerebrovaskulárních příhod řádově o desetinu, nikoli však ischemických komplikací v jiné lokalizaci. Kombinace ASA s dipyridamolem je stejně účinná v sekundární prevenci mozkových příhod jako monoterapie klopidogrelem, je však zatížena mírně vyšší incidencí významných krvácivých příhod.
Dalším lékem s protidestičkovým působením na bázi zvýšení nabídky cAMP, tedy lékem stabilizujícím trombocyty, je inhibitor PDE 3A – cilostazol (tab. 2). Tento lék, primárně užívaný k léčbě klaudikací, bohužel zatím není v ČR dostupný. Jeho indikace v sekundární prevenci iktů vychází z výsledků studie CSPS, kde redukoval riziko recidivy příhody o 42 % oproti placebu. I v následné studii CSPS II u nemocných s recentním non-embolickým iktem byla prokázána non-inferiorita cilostazolu ve srovnání s ASA – rekurence iktů po 29 měsících poklesla asi o čtvrtinu [28]. Cilostazol je v současné době prověřován v řadě indikací jako protidestičkový lék, v indikaci profylaxe trombotických příhod u diabetiků (studie CAPPA) nebo v profylaxi trombóz po implantaci stentu je srovnáván s ASA či je podáván v kombinaci. Výsledky více než dvou desítek probíhajících studií se očekávají kolem roku 2015.
Inhibice agregace trombocytů
Agregace trombocytů je posledním krokem destičkové hemostázy. Spočívá ve vzájemné vazbě trombocytů fibrinovými můstky. Po aktivaci GP receptorů IIb/IIIa je vazná sekvence receptorů obsazena bivalentními vaznými proteiny (fibrinem, vWF, fibronektinem aj.) a vzniká primární trombus. Inhibice v tomto místě je výhodná, neboť zde se spojují všechny podněty iniciující agregaci trombocytů (obr. 1d). Druhou významnou předností blokády agregace je i možnost desagregace destiček, tj. rozpuštění čerstvého destičkového trombu. Na druhé straně je nutno uvést, že tento krok přichází až po degranulaci trombocytů a uvolnění výše popsaných působků, přetrvává tak lokální působení primárního trombu na cévní spasmy, zvýšení permeability endotelu apod. Snížení agregace trombocytů inhibicí receptorů IIb/IIIa na všechny podněty globálně vede nepochybně k nejúčinnější inhibici destičkové hemostázy. Nicméně vedle Skylly je přítomna též Charybda, tedy výrazně zvýšené riziko krvácení. Určitou nevýhodou antagonistů GP receptorů IIb/IIIa (s výjimkou dlouhodobě působícího abciximabu s velkou afinitou k receptoru) je tzv. rebound fenomén. Po uvolnění inhibitoru z vazby zůstává receptor aktivní a může tak dojít ke druhé vlně agregace.
Vývoj v oblasti nových nepeptidových antagonistů GP receptoru IIb/IIIa zatím stagnuje. Velká plejáda „nadějných“ perorálně účinných molekul byla sice testována, nicméně buď byla překážkou jejich zavedení nízká účinnost (zpravidla pro výše zmíněný protrombotický rebound fenomén při nízké afinitě k receptoru) či špatná tolerance – vysoký výskyt hemoragických příhod při
Seznam použité literatury
- [1] Databáze ClinicalTrials.gov registry.
- [2] Van Bockstaele F, Holz JB, Revets H, et al. The development of nanobodies for therapeutic applications. Curr Opin Investig Drugs 2009; 10: 1212–1224.
- [3] Ulrichts H, Silence K, Schoolmeester A, et al. Antithrombotic drug candidate ALX-0081 shows superior preclinical efficacy and safety compared with currently marketed antiplatelet drugs. Blood 2011; 118: 757–765.
- [4] Markus HS, McCollum C, Imray C, et al. The von Willebrand inhibitor ARC1779 reduces cerebral embolization after carotid endarterectomy: a randomized trial. Stroke 2011; 42: 2149–2153.
- [5] Jilma-Stohlawetz P, Gorczyca ME, Jilma B, et al. Inhibition of von Willebrand factor by ARC1779 in patients with acute thrombotic thrombocytopenic purpura. Thromb Haemost 2011; 105: 545–552.
- [6] Siller-Matula JM, Merhi Y, Tanguay JF, et al. ARC15105 Is a Potent Antagonist of Von Willebrand Factor Mediated Platelet Activation and Adhesion. Arterioscler Thromb Vasc Biol 2012, 32: 902–909.
- [7] Bültmann A, Li Z, Wagner S, et al. Impact of glycoprotein VI and platelet adhesion on atherosclerosis – a possible role of fibronectin. J Mol Cell Cardiol 2010; 49: 532–542.
- [8] Mega JL, Close SL, Wiviott SD, et al. Genetic variants in ABCB1 and CYP2C19 and cardiovascular outcomes after treatment with clopidogrel and prasugrel. Lancet 2010; 376: 1312–1319.
- [9] Montalescot G, Wiviott SD, Braunwald E, et al. Prasugrel compared with clopidogrel in patients undergoing percutaneous coronary intervention for ST-elevation myocardial infarction (TRITON- TIMI 38): double-blind, randomised controlled trial. Lancet 2009; 373: 723–731.
- [10] HOTLINE I, PLATO favours ticagrelor over clopidogrel in ACS. Eur Heart J 2009; 30: 2541.
- [11] Hildemann SK, Bode C. Improving antiplatelet therapy for atherothrombotic disease: preclinical results with SCH 530348, the first oral thrombin receptor antagonist selective for PAR-1. Hamostaseologie 2009; 29: 349–355.
- [12] Goto S, Yamaguchi T, Ikeda Y, et al. Safety and exploratory efficacy of the novel thrombin receptor (PAR-1) antagonist SCH530348 for non-ST-segment elevation acute coronary syndrome. J Atheroscler Thromb 2010; 17: 156–164.
- [13] Tricoci P and TRACER Investigators. Thrombin-receptor antagonist vorapaxar in acute coronary syndromes. N Engl J Med 2012; 366: 20–33.
- [14] Morrow DA, Scicira BM, Fox KAA, et al. Evaluation of a novel antiplatelet agent for secondary prevention in patients with a history of atherosclerotic disease: design and rationale for the Thrombin-Receptor Antagonist in Secondary Prevention of Atherothrombotic Ischemic Events (TRA 2 degrees P)-TIMI 50 trial. Am Heart J 2009; 158: 335–341.e3.
- [15] O’Donoghue ML and LANCELOT-ACS Investigators. Safety and tolerability of atopaxar in the treatment of patients with acute coronary syndromes: the lessons from antagonizing the cellular effects of Trombin-Acute Coronary Syndromes Trial. Circulation 2011; 123: 1843–1853.
- [16] Wiviott SD and LANCELOT-CAD Investigators. Randomized trial of atopaxar in the treatment of patients with coronary artery disease: the lessons from antagonizing the cellular effect of Trombin-Coronary Artery Disease Trial. Circulation 2011; 123: 1854–1863.
- [17] Charlot M, Grove EL, Hansen PR, et al. Proton pump inhibitor use and risk of adverse cardiovascular events in aspirin treated patients with first time myocardial infarction: nationwide propensity score matched study. BMJ 2011; 342: d2690.
- [18] Lansberg MG, O´Donnell MJ, Khatri P, et al. Antithrombotic and Thrombolytic Therapy for Ischemic Stroke: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141: e601S–36S.
- [19] Costa J, Ferro JM, Matias-Guiu J, et al. Triflusal for preventing serious vascular events in people at high risk. Cochrane Database Syst Rev 2005; doi: 10.1002/14651858. CD004296.pub2
- [20] Bousser MG and PERFORM Study Investigators. Terutroban versus aspirin in patients with cerebral ischaemic events (PERFORM): a randomised, double-blind, parallel-group trial. Lancet 2011; 377: 2013–2022.
- [21] Weitz JI, Eikelboom JW, Samama MM, et al. New Antithrombotic Drugs: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141: e120S–151S.
- [22] Serneri GGN, Coccheri S, Marubini E, et al. Picotamide, a combined inhibitor of thromboxane A2 synthase and receptor, reduces 2-year mortality in diabetics with peripheral arterial disease: the DAVID study. Eur Heart J 2004; 25: 1845–1852.
- [23] Shikata C, Nemoto M, Ebisawa T, et al. Effect of sarpogrelate on cardiovascular disorders. Exp Clin Cardiol 2011; 16: 75–76.
- [24] Higashi and SEASON Investigators. Study design of SEASON registry: prospective Surveillance of cardiovascular Events in an Antiplatelet-treated arterioSclerosis Obliterans patients in Japan. Int Heart J 2010; 51: 337–342.
- [25] Shinohara Y, Nishimaru K, Sawada T, et al. Sarpogrelate-Aspirin Comparative Clinical Study for Efficacy and Safety in Secondary Prevention of Cerebral Infarction (S-ACCESS): a randomized, double-blind, aspirin-controlled trial. Stroke 2008; 39: 1827–1833.
- [26] Kajiwara I, Soejima H, Miyamoto S, et al. Effects of additional treatment of sarpogrelate to aspirin therapy on platelet aggregation and plasma plasminogen activator inhibitor activity in pa-tients with stable effort angina. Thromb Res 2011; 128: 547–551.
- [27] De Schryver E, Algra A, van Gijn J. Dipyridamole for preventing stroke and other vascular events in patients with vascular disease. Cochrane Database of Systematic Reviews 2007, Issue 3. Art. No.: CD001820.
- [28] Shinohara Y, Katayama Y, Uchiyama S, et al. Cilostazol for prevention of secondary stroke (CSPS 2): an aspirin-controlled, double-blind, randomised non-inferiority trial. Lancet Neurol 2010; 9: 959–968.
- [29] Zeymer U, Zahn R. Glycoprotein IIb/IIIa antagonists: new developments. Hamostaseologie 2009; 29: 334–337.
- [30] Hechler B, Freund M, Alame G, et al. The antithrombotic activity of EP224283, a neutralizable dual factor Xa inhibitor/glycoprotein IIbIIIa antagonist, exceeds that of the coadministered parent compounds. J Pharmacol Exp Ther 2011; 338: 412–420.
- [31] Wiedemann D, Schneeberger S, Friedl P, et al. The fibrin-derived peptide Bbeta(15-42) significantly attenuates ischemia-reperfusion injury in a cardiac transplant model. Transplantation 2010;89: 824–829.
- [32] Hallén J, Petzelbauer P, Schwitter J, et al. Impact of time to therapy and presence of collaterals on the efficacy of FX06 in acute ST elevation myocardial infarction: a substudy of the F.I.R.E., the Efficacy of FX06 in the prevention of myocardial reperfusion injury trial. EuroIntervention 2010; 5: 946–952.
- [33] Ohman J, Kudira R, Albinsson S, et al. Ticagrelor induces adenosine triphosphate release from human red blood cells. Biochemical and Biophysical Research Communications 2012; 418: 754–758.