Inhibitory EGFR a jejich porovnání z hlediska lékových interakcí
Souhrn:
Mezi inhibitory receptoru pro epidermální růstový faktor (epidermal growth factor receptor, EGFR) používané při terapii pacientů s pokročilým nemalobuněčným karcinomem plic patří v České republice registrované gefitinib, erlotinib, afatinib a osimertinib. Tato léčiva mohou vyvolávat řadu lékových interakcí různého klinického významu. Rozsah a závažnost lékových interakcí jsou dány vlastnostmi jednotlivých léčiv z hlediska jejich biotransformace, transportu a rozpustnosti ve vodě při pH vyšším než 6. S pomocí údajů obsažených v Databázi lékových interakcí DrugAgency a dalších informací publikovaných v odborné literatuře v letech 2002–2017 bylo prokázáno, že nejvyšší interakční potenciál, tj. schopnost vyvolávat klinicky významné lékové interakce, má erlotinib, mírně menší interakční potenciál pak gefitinib a významně nižší interakční potenciál má osimertinib a zejména afatinib. Ten také lze z hlediska lékových interakcí považovat za nejbezpečnější inhibitor EGFR.
Key words: gefitinib – erlotinib – afatinib – osimertinib – drug interactions – CYP3A4 – CYP2D6 – CYP1A1 – CYP1A2 – OATP1A2 – OATP1B1 – BCRP – P‑glycoprotein – UGT1A1 – proton pump inhibitors – statins – rosuvastatin – strong inhibitors – strong inducers.
Summary:
Inhibitors of epidermal growth factor receptor used in the therapy of patients with advanced non‑small‑cell lung carcinoma include gefitinib, erlotinib, afatinib and osimertinib approved in the Czech Republic. These drugs can cause a range of drug interactions of variable clinical significance. The extent and severity of drug interactions are defined by characteristics of the individual drugs, regarding their biotransformation, transport and solubility in water while pH is higher than 6. Based on the data in the drug interaction database DrugAgency and further information published in medical literature in 2002–2017, it has been confirmed that the highest interaction potential, i.e., the ability to cause clinically relevant drug interactions, is associated with erlotinib and gefitinib has slightly lower interaction potential whereas osimertinib and particularly afatinib show significantly lower interaction potential. With respect to drug interactions, afatinib can be considered as the safest EGFR inhibitor.
Úvod
Mezi inhibitory receptoru pro
epidermální růstový faktor (EGFR, u lidí označovaného též
jako HER1) patří v České republice registrované přípravky
gefitinib, erlotinib, afatinib a osimertinib. Za objev
epidermálního růstového faktoru (EGF) a jeho receptoru byla
v roce 1986 udělena Nobelova cena biochemikovi Stanleymu
Cohenovi (spolu s Ritou Levi Montalcini, která se
zasloužila o objev nervového růstového faktoru) [1].
Inhibitory EGFR nalezly významné praktické uplatnění při
terapii nemalobuněčného karcinomu plic (non small cell lung
cancer, NSCLC) [2‒4]. Základní údaje o inhibitorech EGFR
jsou uvedeny v tabulce 1.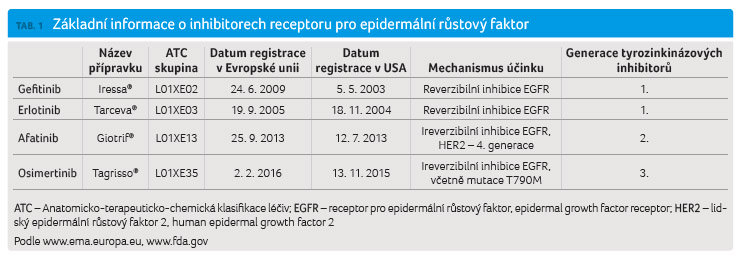
Chemicky se jedná o tzv. malé
molekuly inhibitorů tyrozinkináz s molekulovou hmotností mezi
400‒500 Da. Osimertinib [5] je substituovaný pyrimidin, zatímco
gefitinib [6], erlotinib [7] a afatinib [8] jsou substituované
chinazoliny (obr. 1).
Afatinib se od gefitinibu a erlotinibu významně liší
substitucí, což je příčinou zásadní odlišnosti
biotransfo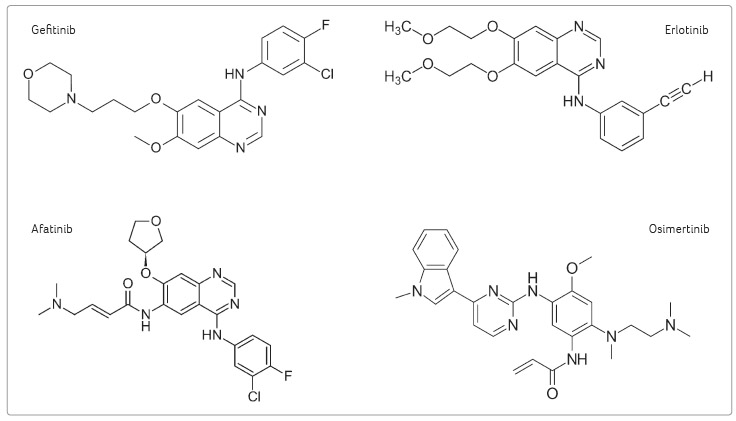 rmace příslušných léčiv a tím i spektra
a závažnosti lékových interakcí.
rmace příslušných léčiv a tím i spektra
a závažnosti lékových interakcí.
Přítomnost 2 enamidové skupiny v molekule afatinibu a osimertinibu zajišťuje ireverzibilní vazbu těchto léčiv na EGFR (receptory rodiny ErbB) a současně je jedním z důvodů nízkého interakčního potenciálu obou léčiv (obr. 2).
Všechny inhibitory EGFR se podávají
![inhibitory eGfr a jejich porovnání z hlediska lékových interakcí 353 kliniCká FArMAkoloGie A FArMACie farmakoLoGie pro kLiNika 41 hodin [6], erlotinib 36 hodin [7], afatinib 37 hodin [8] a osimertinib 48 hodin [5]. Základní vlastnosti inhibitorů EGFR shrnuje tabulka 2. Gefitinib se po perorálním podání extenzivně biotransformuje (> 96 %) za účasti cytochromu P450 3A4 (CYP3A4) (v enterocytu i hepatocytu) a CYP3A5, méně pak za účasti CYP2D6 [6,10]. Gefi tinib není relevantním substrátem efl uxního transportního systému BCRP (protein mnohočetné lékové rezistence, breast cancer resistance protein), je málo citlivým substrátem Pglykoproteinu a infl uxního transportního systému OATP1B1/3 (transportní molekula v bazolaterální membráně hepatocytu, organic anion transporting polypeptide). Na biologickou dostupnost gefi tinibu má významný vliv pH obsahu žaludku. Rozpustnost gefi tinibu ve vodě je závislá na hodnotě pH [6], léčiva zvyšující pH žaludečního obsahu zásadním způsobem snižují expozici gefi tinibu. Léčivá látka neprodlužuje interval QT [10]. Uvedené vlastnosti předurčují významný interakční potenciál gefi tinibu. Erlotinib se po perorálním podání extenzivně biotransformuje (> 98 %) za účasti CYP3A4 (v enterocytu i hepatocytu) a CYP3A5, méně pak za účasti CYP1A2 a CYP2D6, k biotransformaci erlotinibu přispívá též CYP1A1 exprimovaný plicní tkání a CYP1B1 exprimovaný nádorovou tkání [7,11]. Vedle toho je erlotinib substrátem efl uxního transportního systému BCRP (méně pak Pglykoproteinu) a infl ux ního transportního systému OATP1A2 a OATP1B1/3. Erlotinib se vstřebává v tenkém střevě zčásti aktivním transportem pomocí OATP1A2. Inhibice OATP1A2 látkami obsaženými v zeleném čaji (epigalokatechin galát) má za následek snížení expozice erlotinibu o 70 %, které je zatím prokázané v neklinických studiích u potka nů [14]. Rozpustnost erlotinibu ve vodě je závislá na hodnotě pH [7], léčiva zvyšující pH žaludečního obsahu zásadním způsobem snižují expozici erlotinibu. Rovněž kouření významně snižuje expozici erlotinibu [15]. Erlotinib neprodlužuje interval QT. Uvedené vlastnosti předurčují velmi vysoký interakční potenciál léčiva. Afatinib se po perorálním podání jen minimálně biotransformuje (< 10 %). Jeho minimální biotransformace je zprostředkována CYP3A4 a fl avinmonooxygenázou 3 (fl avincontaining monooxygenase 3, FMO3) [8,12]. Afatinib je substrátem efl uxního transportního systému BCRP a Pglykoproteinu. Není však substrátem infl uxního transportního systému OATP1A2 ani OATP1B1/3. Acidita obsahu žaludku nemá vliv na biologickou dostupnost afatinibu [8]. Afatinib neprodlužuje interval QT, afatinib má jen minimum lékových interakcí, z nichž žádná není klinicky významná. Osimertinib se po perorálním podání extenzivně biotransformuje (> 98 %) za účasti CYP3A4 (v enterocytu i hepatocytu) a CYP3A5 [5,13]. Osimertinib je substrátem efl uxního transportního systému BCRP a Pglykoproteinu. Není však substrátem infl uxního transportního systému OATP1A2 ani OATP1B1/3. Acidita obsahu žaludku nemá vliv na biologickou dostupnost osimertinibu [5]. Osimertinib může prodloužit interval QT a vést k závažným komorovým arytmiím [13]. Léčivo má jen málo lékových interakcí, některé z nich však jsou klinicky významné. OBR. 1 Chemická struktura inhibitorů receptoru pro epidermální růstový faktor; podle Wikipedia, www.wikipedia.org. Gefi tinib erlotinib afatinib osimertinib OBR. 2 způsob ireverzibilní vazby afatinibu a osimertinibu; upraveno podle [9] – Wikipedia. Cys – cystein; eGFr – receptor pro epidermální růstový faktor, epidermal growth factor receptor](https://www.remedia.cz/photo-a-31247---.jpg) perorálně v jedné denní dávce, protože mají dlouhý
biologický poločas ‒ gefitinib 41 hodin [6], erlotinib 36 hodin
[7], afatinib 37 hodin [8] a osimertinib 48 hodin [5].
Základní vlastnosti inhibitorů EGFR shrnuje tabulka 2.
perorálně v jedné denní dávce, protože mají dlouhý
biologický poločas ‒ gefitinib 41 hodin [6], erlotinib 36 hodin
[7], afatinib 37 hodin [8] a osimertinib 48 hodin [5].
Základní vlastnosti inhibitorů EGFR shrnuje tabulka 2.
Gefitinib se
po perorálním podání extenzivně biotransformuje (> 96 %)
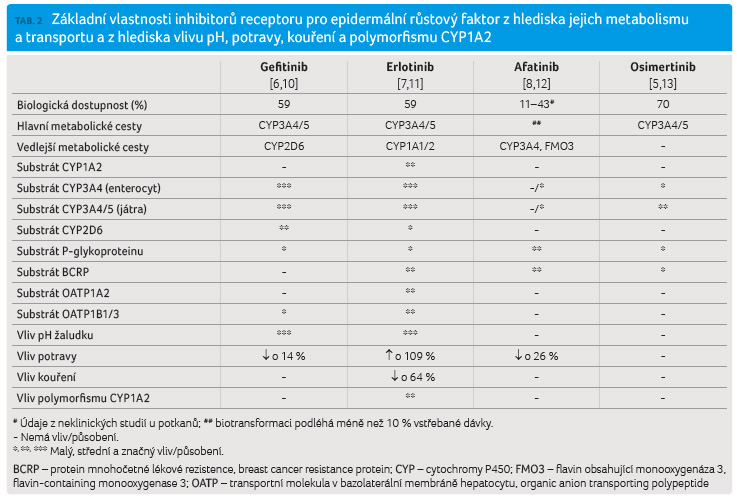 za účasti cytochromu P450 3A4 (CYP3A4) (v enterocytu
i hepatocytu) a CYP3A5, méně pak za účasti CYP2D6
[6,10]. Gefitinib není relevantním substrátem efluxního
transportního systému BCRP (protein mnohočetné lékové
rezistence, breast cancer resistance protein), je málo citlivým
substrátem P glykoproteinu a influxního transportního
systému OATP1B1/3 (transportní molekula v bazolaterální
membráně hepatocytu, organic anion transporting polypeptide).
Na biologickou dostupnost gefitinibu má významný vliv pH
obsahu žaludku. Rozpustnost gefitinibu ve vodě je závislá na
hodnotě pH [6], léčiva zvyšující pH žaludečního obsahu
zásadním způsobem snižují expozici gefitinibu. Léčivá látka
neprodlužuje interval QT [10]. Uvedené vlastnosti předurčují
významný interakční potenciál gefitinibu.
za účasti cytochromu P450 3A4 (CYP3A4) (v enterocytu
i hepatocytu) a CYP3A5, méně pak za účasti CYP2D6
[6,10]. Gefitinib není relevantním substrátem efluxního
transportního systému BCRP (protein mnohočetné lékové
rezistence, breast cancer resistance protein), je málo citlivým
substrátem P glykoproteinu a influxního transportního
systému OATP1B1/3 (transportní molekula v bazolaterální
membráně hepatocytu, organic anion transporting polypeptide).
Na biologickou dostupnost gefitinibu má významný vliv pH
obsahu žaludku. Rozpustnost gefitinibu ve vodě je závislá na
hodnotě pH [6], léčiva zvyšující pH žaludečního obsahu
zásadním způsobem snižují expozici gefitinibu. Léčivá látka
neprodlužuje interval QT [10]. Uvedené vlastnosti předurčují
významný interakční potenciál gefitinibu.
Erlotinib se po perorálním podání extenzivně biotransformuje (> 98 %) za účasti CYP3A4 (v enterocytu i hepatocytu) a CYP3A5, méně pak za účasti CYP1A2 a CYP2D6, k biotransformaci erlotinibu přispívá též CYP1A1 exprimovaný plicní tkání a CYP1B1 exprimovaný nádorovou tkání [7,11]. Vedle toho je erlotinib substrátem efluxního transportního systému BCRP (méně pak P glykoproteinu) a influxního transportního systému OATP1A2 a OATP1B1/3. Erlotinib se vstřebává v tenkém střevě zčásti aktivním transportem pomocí OATP1A2. Inhibice OATP1A2 látkami obsaženými v zeleném čaji (epigalokatechin galát) má za následek snížení expozice erlotinibu o 70 %, které je zatím prokázané v neklinických studiích u potkanů [14]. Rozpustnost erlotinibu ve vodě je závislá na hodnotě pH [7], léčiva zvyšující pH žaludečního obsahu zásadním způsobem snižují expozici erlotinibu. Rovněž kouření významně snižuje expozici erlotinibu [15]. Erlotinib neprodlužuje interval QT. Uvedené vlastnosti předurčují velmi vysoký interakční potenciál léčiva.
Afatinib se po perorálním podání jen minimálně biotransformuje (< 10 %). Jeho minimální biotransformace je zprostředkována CYP3A4 a flavin monooxygenázou 3 (flavin containing monooxygenase 3, FMO3) [8,12]. Afatinib je substrátem efluxního transportního systému BCRP a P glykoproteinu. Není však substrátem influxního transportního systému OATP1A2 ani OATP1B1/3. Acidita obsahu žaludku nemá vliv na biologickou dostupnost afatinibu [8]. Afatinib neprodlužuje interval QT, afatinib má jen minimum lékových interakcí, z nichž žádná není klinicky významná.
Osimertinib se po perorálním podání extenzivně biotransformuje (> 98 %) za účasti CYP3A4 (v enterocytu i hepatocytu) a CYP3A5 [5,13]. Osimertinib je substrátem efluxního transportního systému BCRP a P glykoproteinu. Není však substrátem influxního transportního systému OATP1A2 ani OATP1B1/3. Acidita obsahu žaludku nemá vliv na biologickou dostupnost osimertinibu [5]. Osimertinib může prodloužit interval QT a vést k závažným komorovým arytmiím [13]. Léčivo má jen málo lékových interakcí, některé z nich však jsou klinicky významné.
Lékové interakce inhibitorů EGFR
Díky chemické struktuře a odlišným vlastnostem jednotlivých inhibitorů EGFR mají tato léčiva odlišné spektrum a odlišnou závažnost lékových interakcí. Gefitinib a erlotinib jsou citlivé substráty CYP3A4/5 [6,7], méně citlivým substrátem CYP3A4/5 je osimertinib [5], afatinib není relevantním substrátem CYP3A4 [8]. Gefitinib, erlotinib, afatinib a osimertinib jsou substráty P glykoproteinu a erlotinib, afatinib a osimertinib jsou substráty BCRP [5‒8]. Gefitinib a erlotinib jsou navíc substráty transportního systému OATP1B1/3 a erlotinib dále pak též substrátem OATP1A2 [14].
Lékové interakce inhibitorů EGFR ve smyslu ovlivnění metabolismu nebo transportu těchto léčiv tedy lze očekávat jak s inhibitory CYP3A4, tak i s induktory CYP3A4, v obou případech zejména silnými. Vedle toho lze očekávat lékové interakce s inhibitory BCRP a s inhibitory či induktory P glykoproteinu. Pro klinickou praxi je významné, že většina uvedených interakcí byla cílenými studiemi dobře doložena, resp. bylo v některých případech doloženo, že příslušná léková interakce nemá klinický význam. V případech, kdy bylo zjištěno, že se jedná o klinicky významnou lékovou interakci, byla navržena relevantní klinická doporučení. Některá z těchto doporučení však nevycházejí z přímých důkazů, ale z analogií zjištěných u jiných léčivých přípravků nebo z farmakokinetických modelování.
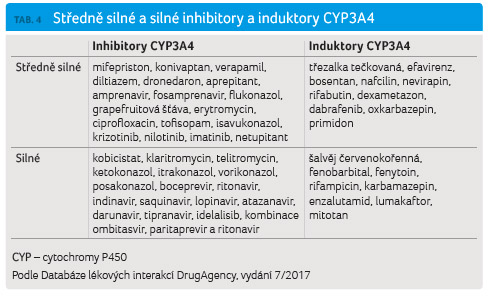
Lékové interakce se silnými inhibitory nebo induktory CYP3A4 a P glykoproteinu
Se všemi inhibitory EGFR byly provedeny cílené studie zaměřené na průkaz lékových interakcí s ketokonazolem, resp. itrakonazolem jako silnými inhibitory CYP3A4 (a současně středně silnými inhibitory P glykoproteinu). V případě afatinibu byla provedena studie s ritonavirem, který je též silným inhibitorem CYP3A4 a středně silným inhibitorem P glykoproteinu. Současně byly provedeny cílené studie zaměřené na průkaz lékových interakcí s rifampicinem jako silným induktorem CYP3A4 a silným induktorem P glykoproteinu.
Z tabulky 3 jednoznačně vyplývá, že všechny substráty CYP3A4
interagují se silnými inhibitory, respektive induktory
CYP3A4/P glykoproteinu (jejich přehled přinášejí tab. 4 a 5),
přičemž v případě induktorů se jedná o klinicky
významné lékové interakce u gefitinibu, erlotinibu
a osimertinibu a v případě inhibitorů se jedná
o klinicky významné lékové interakce u gefitinibu
a erlotinibu.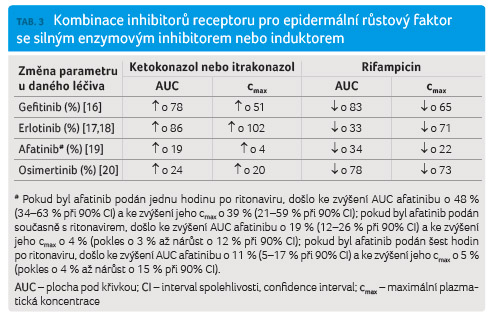
Výrobce gefitinibu v ČR
(v Evropské unii, EU) pro současné užívání silných
inhibitorů CYP3A4 uvádí [10]: „U pacientů
s genotypem pomalých metabolizátorů pro substráty CYP2D6
může vést léčba účinnými inhibitory CYP3A4 ke zvýšení
plazmatických koncentrací gefitinibu. Při zahájení léčby
inhibitory CYP3A4 by měli být pacienti pečlivě sledováni
s ohledem na nežádoucí účinky gefitinibu.“ Tato
informace není zcela přesná, neboť současná léčba gefitinibem
a účinnými inhibitory CYP3A4 vede ke zvýšení expozice
gefitinibu i u rychlých metabolizátorů CYP2D6, i když
v případě pomalých nepochybně dojde k blokádě obou
cest metabolizace a k nejvyššímu nárůstu expozice
gefitinibu. Výrobce gefitinibu v USA doporučuje monitorování
nežádoucích účinků u všech pacientů [21]. Pro současné
 užívání silných induktorů CYP3A4 uvádí výrobce gefitinibu
v ČR (v EU) následující: „Látky indukující
aktivitu CYP3A4 mohou zvyšovat metabolismus a snižovat
plazmatické koncentrace gefitinibu, a tím snižovat účinnost
gefitinibu. Současné podávání léčivých přípravků, které
indukují aktivitu CYP3A4 (tj. fenytoin, karbamazepin,
rifampicin, barbituráty a bylinné přípravky obsahující
třezalku tečkovanou/Hypericum
perforatum), by mělo být vyloučeno.“
užívání silných induktorů CYP3A4 uvádí výrobce gefitinibu
v ČR (v EU) následující: „Látky indukující
aktivitu CYP3A4 mohou zvyšovat metabolismus a snižovat
plazmatické koncentrace gefitinibu, a tím snižovat účinnost
gefitinibu. Současné podávání léčivých přípravků, které
indukují aktivitu CYP3A4 (tj. fenytoin, karbamazepin,
rifampicin, barbituráty a bylinné přípravky obsahující
třezalku tečkovanou/Hypericum
perforatum), by mělo být vyloučeno.“
Výrobce gefitinibu v USA
doporučuje po dobu užívání silných induktorů
CYP3A4 zvýšit dávky gefitinibu 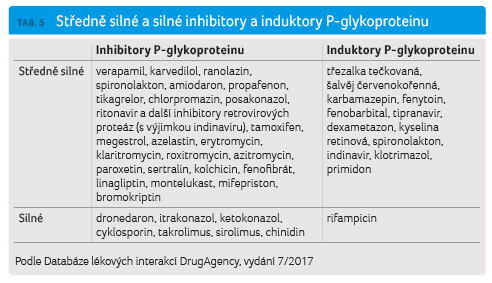 z obvyklých 250 mg
denně na 500 mg denně a 7 dnů po ukončení
podávání silného induktoru CYP3A4 pak dávky gefitinibu snížit
na obvyklých 250 mg denně [21]. Z hlediska medicíny
založené na důkazech (evidence based medicine, EBM) je
třeba uvést, že výše uvedená studie prokazující lékovou
interakci mezi gefitinibem a rifampicinem byla provedena
s dávkou gefitinibu ve výši 500 mg [16]
a farmakokinetické parametry gefitinibu zjištěné v této
studii opravňují k předpokladu, že dávka gefitinibu ve výši
500 mg spolu se silným induktorem povede k obdobné expozici
gefitinibu jako dávka gefitinibu ve výši 250 mg podávaná
bez silného induktoru. Takzvaný on label postup pro ČR (EU)
představuje vyloučení současného podávání gefitinibu
a silných induktorů CYP3A4.
z obvyklých 250 mg
denně na 500 mg denně a 7 dnů po ukončení
podávání silného induktoru CYP3A4 pak dávky gefitinibu snížit
na obvyklých 250 mg denně [21]. Z hlediska medicíny
založené na důkazech (evidence based medicine, EBM) je
třeba uvést, že výše uvedená studie prokazující lékovou
interakci mezi gefitinibem a rifampicinem byla provedena
s dávkou gefitinibu ve výši 500 mg [16]
a farmakokinetické parametry gefitinibu zjištěné v této
studii opravňují k předpokladu, že dávka gefitinibu ve výši
500 mg spolu se silným induktorem povede k obdobné expozici
gefitinibu jako dávka gefitinibu ve výši 250 mg podávaná
bez silného induktoru. Takzvaný on label postup pro ČR (EU)
představuje vyloučení současného podávání gefitinibu
a silných induktorů CYP3A4.
Výrobce erlotinibu v ČR (v EU) výslovně uvádí [11], že při současném užívání substrátů a modulátorů CYP3A4 může být zapotřebí upravit dávkování. Za touto výmluvnou formulací se skrývá doporučení pro současné podávání silných inhibitorů: je třeba postupovat se zvýšenou opatrností při kombinování erlotinibu a silných inhibitorů CYP3A4, např. azolových antimykotik (mj. ketokonazol, itrakonazol, vorikonazol), proteázových inhibitorů, erytromycinu nebo klaritromycinu. V případě nutnosti je možné dávku erlotinibu snížit, zejména pokud dochází k projevům toxicity.
Výrobce erlotinibu v USA uvádí [22], že dávku přípravku je třeba snížit o 50 %, a je li to možné, je třeba se vyvarovat společného podávání silných inhibitorů. Dále pak uvádí doporučení pro současné užívání silných induktorů: po současném podání rifampicinu a jednorázové dávky 450 mg erlotinibu činila průměrná expozice erlotinibu 57,5 % hodnoty naměřené po jednorázovém podání 150 mg erlotinibu bez současného podání rifampicinu. Je proto nutné se vyvarovat současného podání erlotinibu a induktorů CYP3A4. U nemocných, u nichž je nutná souběžná léčba erlotinibem a silnými induktory CYP3A4, jako je například rifampicin, je nutno zvážit zvýšení dávky erlotinibu na 300 mg při současném pečlivém sledování bezpečnosti (včetně sledování ledvinných a jaterních funkcí a elektrolytů v séru). V případě dobré tolerance po dobu delší než dva týdny může být zváženo zvýšení dávky na 450 mg při současném pečlivém sledování bezpečnosti. Výrobce erlotinibu v USA doporučuje zvyšovat dávky erlotinibu o 50 mg denně ve dvoutýdenních intervalech a při dobré toleranci až k dávce 450 mg a opět uvádí, že je li to možné, je třeba se vyvarovat společného podávání silných inhibitorů.
Výrobce afatinibu v ČR (v EU) se k současnému podávání silných inhibitorů nebo induktorů CYP3A4 nevyjadřuje [12], neboť se afatinib cestou CYP3A4 prakticky nebiotransformuje. Je však třeba uvést, že část inhibitorů, resp. induktorů CYP3A4 obdobným způsobem ovlivňuje též P glykoproteinový transportní systém. Řada inhibitorů i induktorů CYP3A4 je současně inhibitorem nebo induktorem P glykoproteinu. Lékové interakce inhibitorů nebo induktorů P glykoproteinu s afatinibem však nejsou klinicky významné. V případě současného podávání inhibitorů je řešením co největší odstup mezi podáním afatinibu a inhibitoru P glykoproteinu, kdy v případě odstupu 12 hodin k lékové interakci již nedochází. Důvodem je plná reverzibilita inhibice P glykoproteinu a její krátké trvání. Výrobce afatinibu v ČR (v EU) proto doporučuje podávat dávku silných inhibitorů P glykoproteinu (včetně ritonaviru, cyklosporinu, ketokonazolu, itrakonazolu, erytromycinu, verapamilu, chinidinu, takrolimu, nelfinaviru, saquinaviru, amiodaronu a dalších) střídavě, nejlépe 6 hodin nebo 12 hodin od podání afatinibu [12]. V případě induktorů (indukce obecně přetrvává 7 a více dnů po ukončení podávání induktoru) výrobce afatinibu v ČR (v EU) pouze konstatuje, že induktory P glykoproteinu mohou snížit expozici afatinibu. Výrobce afatinibu v USA uvádí [23], že v případě užívání silných inhibitorů P glykoproteinu lze při nedostatečné toleranci dávku afatinibu snížit o 10 mg denně a v případě užívání silných induktorů P glykoproteinu lze dávku afatinibu zvýšit o 10 mg denně a užívat ji ještě 2‒3 dny po ukončení podávání induktoru.
Výrobce osimertinibu v ČR (v EU) výslovně uvádí, že současné užívání třezalky tečkované je kontraindikované [13]. Dále doporučuje nepodávat souběžně osimertinib se silnými induktory CYP3A (např. fenytoin, rifampicin a karbamazepin) a uvádí, že středně silné induktory CYP3A4 (např. bosentan, efavirenz, etravirin, modafinil) mohou též snižovat expozici osimertinibu, a mají se proto používat s opatrností nebo zcela vyloučit, pokud je to možné. Nejsou dostupné žádné klinické údaje, na jejichž podkladě by bylo možné doporučit úpravu dávkování osimertinibu [14]. Dále výrobce osimertinibu v ČR (v EU) uvádí, že inhibitory CYP3A4 pravděpodobně neovlivňují expozici osimertinibu. Výrobce osimertinibu v USA naproti tomu uvádí, že není li možné se vyhnout silným induktorům CYP3A4, je třeba zvýšit dávku osimertinibu z obvyklých 80 mg denně na 160 mg denně [24].
Závěrem lze konstatovat, že současné podávání gefitinibu nebo erlotinibu se silnými inhibitory nebo induktory CYP3A4 představuje buď riziko zvýšení výskytu nežádoucích účinků, nebo přerušení či předčasného ukončení terapie (inhibitory), resp. selhání terapie (induktory). Tyto interakce jsou obecně hodnoceny jako závažné a podle výrobců gefitinibu a erlotinibu by mělo být současnému podávání takových kombinací zabráněno [10,11]. Pro systém veřejného zdravotního pojištění představuje kombinace gefitinibu nebo erlotinibu zejména s induktory CYP3A4 zátěž danou buď snížením či selháním účinku, nebo zvýšením dávek nad obvyklé množství (a adekvátně vyšší finanční plnění). V případě citlivých substrátů CYP3A4/P glykoproteinu, jako jsou gefitinib nebo erlotinib, může dojít ke kritické lékové interakci též při použití slabších induktorů CYP3A4, jako je např. extrakt ze ženšenu pravého. Byla popsána kazuistika 36leté pacientky [25], u níž byl diagnostikován NSCLC ve stadiu IV. Bylo zahájeno podávání gefitinibu v dávkách 250 mg denně. Po 9 týdnech terapie si při kontrole pacientka stěžovala na zhoršení dyspnoe a CT vyšetření potvrdilo progresi onemocnění. Pacientce bylo doporučeno ukončení terapie gefitinibem. Nemocná informovala lékaře, že současně s gefitinibem užívala doplněk stravy obsahující ženšen pravý (a jako další složky extrakty z hub troudnatec kopytovitý, rezavec šikmý a ohňovec brázditý). Doplněk stravy tedy přestala užívat a byla nadále léčena pouze gefitinibem v dávkách 250 mg denně. Při kontrole za 4 týdny se její stav zlepšil a pacientka nadále pokračuje v terapii gefitinibem již 30 týdnů.
Lékové interakce se silnými inhibitory nebo induktory CYP1A2
Erlotinib je klinicky relevantní substrát CYP1A2. Sám však není ani inhibitorem, ani induktorem tohoto izoenzymu cytochromu P450 [7]. Ve studii u 28 zdravých dobrovolníků [26] ve věku 19‒53 let byl podáván ciprofloxacin v dávkách 750 mg dvakrát denně (jako silný inhibitor CYP1A2) po dobu 5 dnů, před zahájením podávání ciprofloxacinu a spolu s jeho poslední dávkou byla podána jednorázová dávka erlotinibu ve výši 100 mg. Došlo ke zvýšení plochy pod křivkou (AUC) erlotinibu o 39 % (24‒56 % při 90% CI [interval spolehlivosti]), ke zvýšení AUC aktivního metabolitu erlotinibu o 60 % (40‒87 % při 90% CI) a ke zvýšení jeho maximálních plazmatických koncentrací (cmax) o 48 % (20‒82 % při 90% CI). Byly též popsány kazuistiky dvou pacientů [27] ve věku 60 a 79 let léčených erlotinibem po dobu 3 měsíců, resp. 6 měsíců, u nichž došlo během terapie erlotinibem k perforaci trávicího ústrojí. Jako rizikový faktor byly identifikovány ciprofloxacin, který zvyšuje expozici erlotinibu, a současné podávání glukokortikoidů, jež zvyšuje riziko perforace trávicího ústrojí. Oba pacienti zemřeli. Výrobce erlotinibu v ČR (v EU) uvádí [11], že při kombinaci erlotinibu s ciprofloxacinem nebo jiným silným inhibitorem CYP1A2 (např. fluvoxamin) je třeba postupovat se zvýšenou opatrností. Pokud se vyskytnou nežádoucí účinky erlotinibu, jeho dávka může být snížena. Výrobce erlotinibu v USA pak uvádí [22], že snížení by mělo být o 50 mg denně.
Značný problém může představovat kouření během terapie erlotinibem. Cigaretový kouř obsahuje polykondenzované aromatické uhlovodíky a řadu dalších látek, které působí jako středně silné až silné induktory CYP1A1 a CYP1A2 [28,29] (tab. 6). V cílené farmakokinetické studii bylo u zdravých dobrovolníků prokázáno, že kouření vedlo ke snížení expozice jednorázové dávce erlotinibu o 64 % (46‒76 % při 95% CI) [15]. Výrobce erlotinibu v ČR uvádí [11]: „Kuřákům je třeba doporučit přerušit kouření, protože jsou u nich plazmatické koncentrace erlotinibu nižší než u nekuřáků.“ Toto snížení je pravděpodobně klinicky významné. Výrobce erlotinibu dále uvádí: „Ve studii fáze III byla minimální plazmatická koncentrace erlotinibu v ustáleném stavu, které bylo dosaženo u stávajících kuřáků, 0,65 µg/ml (n = 16), což bylo přibližně dvakrát méně než u bývalých kuřáků či nemocných, kteří nikdy nekouřili (1,28 µg/ml, n = 108).“ Ve studii fáze I s eskalací dávky u nemocných s NSCLC, kteří byli stávajícími kuřáky, ukazovaly farmakokinetické analýzy v ustáleném stavu proporcionální vzestup expozice erlotinibu, pokud byla dávka erlotinibu zvýšena ze 150 mg na maximální tolerovanou dávku 300 mg. Minimální plazmatická koncentrace v ustáleném stavu dosáhla u kuřáků při dávce 300 mg hodnoty 1,22 µg/ml. V této souvislosti je zajímavé, že v podmínkách úhrady erlotinibu ze systému veřejného zdravotního pojištění není o zákazu souběžného kouření uvedeno nic, zatímco v jiných případech bývá kouření důvodem pro nehrazení léčiva.
Lékové interakce se silnými inhibitory CYP2D6
Gefitinib je klinicky relevantní substrát CYP2D6 a současně je slabým inhibitorem CYP2D6 [6]. Ačkoliv dosud nebyla provedena žádná cílená klinická studie prokazující lékovou interakci mezi silnými inhibitory CYP2D6 a gefitinibem, je ze studií zaměřených na generický polymorfismus známo, že u pomalých metabolizátorů CYP2D6 dochází ke zvýšení expozice gefitinibu (přibližně dvojnásobnému) [10]. Současně dochází ke snížení expozice hlavnímu metabolitu (O desmetylgefitinibu) v poměru 1,41 : 0,86 : 0,24 u rychlých, intermediárních a pomalých metabolizátorů CYP2D6 [30].
Zvýšení expozice gefitinibu
u pomalých metabolizátorů CYP2D6 je klinicky významné nejen
z důvodu změny farmakokinetických parametrů, ale
pravděpodobně též z důvodu toxicity, např. kožní [31]
nebo jaterní [32], a to zejména při současné terapii
inhibitory CYP3A4 [10] (blokáda obou cest metabolizace). Lze tedy
důvodně očekávat, že současné podávání gefitinibu se
silnými inhibitory CYP2D6 bude spojeno se zvýšením expozice
gefitinibu a pravděpodobně se  zvýšením jeho toxicity.
Souběžný výskyt NSCLC a deprese byl měl vést k úvaze,
že některá antidepresiva jsou silnými inhibitory CYP2D6
(fluoxetin, paroxetin, bupropion) a jiná pak středně silnými
inhibitory CYP2D6 (fluvoxamin, sertralin, moklobemid, duloxetin),
a proto je vhodné volit jinou antidepresivní terapii (tab. 7).
zvýšením jeho toxicity.
Souběžný výskyt NSCLC a deprese byl měl vést k úvaze,
že některá antidepresiva jsou silnými inhibitory CYP2D6
(fluoxetin, paroxetin, bupropion) a jiná pak středně silnými
inhibitory CYP2D6 (fluvoxamin, sertralin, moklobemid, duloxetin),
a proto je vhodné volit jinou antidepresivní terapii (tab. 7).
Lékové interakce s protisekrečními léky
Řada inhibitorů tyrozinkináz je
rozpustná ve vodě pouze při nízkých hodnotách pH. Zvýšením
hodnoty pH (pH > 5) se rozpustnost těchto léčiv
prudce snižuje, což obvykle vede k poklesu jejich biologické
dostupnosti (tab. 8).
In vivo byl negativní
vliv suprese kyselé žaludeční sekrece na biologickou
dostupnost prokázán u bosutinibu, kabozantinibu, dasatinibu,
erlotinibu, gefitinibu, lapatinibu a pazopanibu, in
vitro pak byl prokázán u dabrafenibu a ibrutinibu
[33].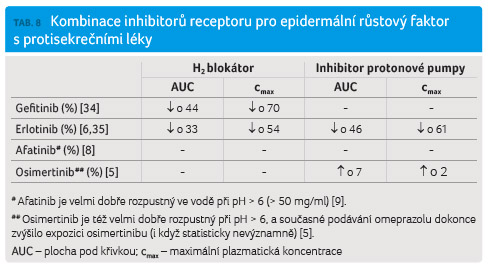
Podání gefitinibu současně s H2 blokátorem (a velmi pravděpodobně také s inhibitorem protonové pumpy) vede ke snížení cmax gefitinibu o 70 % (a ke snížení biologické dostupnosti o 44 %) [34]. V případě gefitinibu nejsou důkazy o vlivu současného podávání protisekrečních léčiv na prognózu nemocných.
Podání erlotinibu současně s inhibitorem protonové pumpy či H2 blokátorem vede ke snížení cmax erlotinibu o 61 % (a ke snížení biologické dostupnosti o 46 %) [35]. Bylo prokázáno, že současné užívání protisekrečních léčiv zkracuje dobu do progrese onemocnění. Relativní riziko snížení terapeutické odpovědi vyjádřené jako čas do progrese onemocnění bylo 1,83 (95% CI 1,48–2,25), resp. vyjádřené jako celkové přežití bylo 1,37 (95% CI 1,11–1,69) v neprospěch pacientů užívajících protisekreční léčiva [36]. Výrobce erlotinibu v ČR uvádí [11]: „Pro erlotinib je charakteristická snížená rozpustnost při hodnotě pH vyšší než 5. Léčivé přípravky, které ovlivňují pH v horní části trávicího traktu, jako jsou inhibitory protonové pumpy, H2 antagonisté a antacida, mohou ovlivnit rozpustnost erlotinibu a tím jeho biologickou dostupnost.“ Není pravděpodobné, že by zvýšení dávky erlotinibu při současném podávání takových léků mohlo kompenzovat ztrátu expozice. Kombinace erlotinibu s inhibitory protonové pumpy by se neměla užívat. K zajímavým zjištěním, s potenciálním využitím při řešení lékových interakcí erlotinibu a protisekrečních léčiv, dospěli nizozemští autoři. V randomizované studii u 28 pacientů s NSCLC podávali erlotinib souběžně s esomeprazolem po dobu 7 dnů, nebo podávali samotný erlotinib a 7. den studie byla podána buď coca cola, nebo voda v množství 250 ml [37]. V případě podání nápoje coca cola pacientům užívajícím souběžně erlotinib a esomeprazol došlo ke zvýšení expozice erlotinibu průměrně o 39 % (pokles o 12 % až nárůst o 136 % při 90% CI), současně došlo ke zvýšení cmax erlotinibu průměrně o 42 % (pokles o 4 % až nárůst o 199 % při 90% CI). V případě pacientů léčených pouze erlotinibem došlo ke zvýšení expozice erlotinibu pouze o 9 % (pokles o 10 % až nárůst o 30 % při 90% CI), v tomto případě se výše cmax nezměnila.
Vzhledem k dobré rozpustnosti afatinibu při pH > 6 nebylo výrobcem přikročeno k provedení farmakokinetické studie cílené na průkaz lékové interakce [8]. Taková studie však byla provedena v případě hodnoty pH > 6 dobře rozpustného osimertinibu, a to s negativním výsledkem ‒ omeprazol neměl vliv na biologickou dostupnost osimertinibu [38].
Lékové interakce s citlivými substráty enzymů nebo transportních systémů
Gefitinib je in vitro slabým inhibitorem CYP2D6. In vivo byla tato aktivita prokázána ve studii u 18 pacientů se solidními tumory ve věku 44‒66 let, z nichž tři studii nedokončili (jeden kvůli souběžné aplikaci klaritromycinu a dva kvůli nutnému snížení dávkování gefitinibu z důvodu vzniku exantému) [16]. Pacientům byl podáván gefitinib v dávkách 500 mg jednou denně po dobu 28 dnů, před zahájením podávání gefitinibu a 15. den jeho užívání byla podána jednorázová dávka metoprololu ve výši 50 mg. Došlo ke zvýšení AUC metoprololu o 35 % a ke zvýšení jeho cmax o 10 %. Léková interakce není hodnocena jako klinicky významná, avšak v případě některých jiných citlivých substrátů CYP2D6 s úzkým terapeutickým oknem nelze vyloučit závažnější lékovou interakci. Gefitinib je dále in vitro slabým induktorem CYP3A4 a P glykoproteinu. To však patrně nemá žádné dopady z hlediska lékových interakcí.
Erlotinib je in vitro silným inhibitorem CYP1A1 a uridindifosfát glukuronyltransferázy 1A1 (UGT1A1) [7]. Inhibice CYP1A1 je vzhledem k minimální expresi tohoto izoenzymu v lidských buňkách patrně bez dopadu na lékové interakce. Inhibice UGT1A1 by mohla mít vliv na biotransformaci některých léčiv, která se glukuronizují prostřednictvím tohoto izoenzymu (např. irinotekan). To také bylo prokázáno ve studii s použitím lidských hepatocytů. V této studii bylo zjištěno, že erlotinib je silným inhibitorem UGT1A1, přičemž bylo odhadnuto, že dávka erlotinibu 50 mg nebo vyšší může zvýšit expozici aktivnímu metabolitu irinotekanu (SN 38) o 24 % [39]. Erlotinib je dále in vitro slabým induktorem CYP3A4 a P glykoproteinu [7]. To však patrně nemá žádné dopady z hlediska lékových interakcí.
Afatinib je in vitro slabým inhibitorem BCRP a P glykoproteinu [8]. V případě P glykoproteinu je míra inhibice nízká a nepředpokládá se, že by mohlo současné podávání afatinibu se substráty P glykoproteinu vést ke klinicky závažným lékovým interakcím. V případě inhibice BCRP výrobce afatinibu [12] připouští možnost lékových interakcí afatinibu se substráty BCRP (jako jsou rosuvastatin nebo sulfasalazin), žádné cílené klinické studie však dosud nebyly provedeny.
Osimertinib je (podobně jako afatinib)
in vitro inhibitorem
BRCP [5]. Byla provedena cílená studie prokazující lékovou
interakci mezi osimertinibem a rosuvastatinem (jako substrátem
BCRP). V této studii u 44 pacientů s NSCLC
s prokázanou mutací EGFR, průměrného věku 61,5 roku, byl
podáván osimertinib v dávkách 80 mg jednou denně po dobu
31 dnů, před zahájením podávání osimertinibu a v 29. dni
jeho užívání byla podána jednorázová dávka rosuvastatinu
ve výši 20 mg. Vlivem osimertinibu došlo ke zvýšení
AUC rosuvastatinu o 35 % (15‒57 % při 90% CI)
a ke zvýšení jeho cmax o 72 %
(46‒103 % při 90% CI) [40]. Tato léková interakce je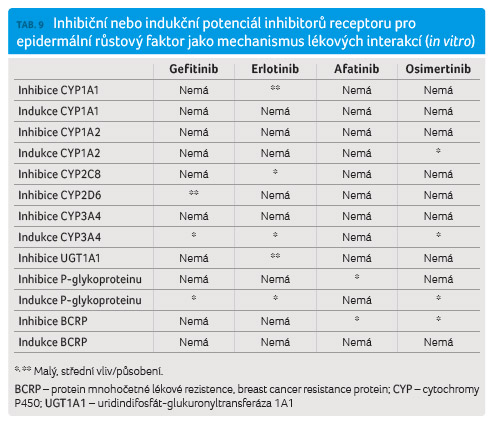 hodnocena
jako středně závažná. Osimertinib je vedle toho in
vitro slabým induktorem CYP1A2, CYP3A4 a P glykoproteinu
[5]. To vysvětluje mírné snížení expozice simvastatinu
prokázané v cílené klinické studii. V této studii
u 52 pacientů s NSCLC s prokázanou mutací EGFR,
průměrného věku 63,5 roku, z nichž pro srovnání farmakokinetiky simvastatinu bylo hodnotitelných 41, byl podáván
osimertinib v dávkách 80 mg jednou denně po dobu 30
dnů, před zahájením podávání osimertinibu a v 29. dni
jeho užívání byla podána jednorázová dávka simvastatinu
ve výši 40 mg [41]. Vlivem osimertinibu došlo ke snížení
AUC simvastatinu o 9 % (od zvýšení o 8 %
po snížení o 23 % při 90% CI) a ke snížení
jeho cmax o 23 % (6‒37 % při 90% CI). Inhibiční nebo indukční potenciál
inhibitorů EGFR shrnuje tabulka 9.
hodnocena
jako středně závažná. Osimertinib je vedle toho in
vitro slabým induktorem CYP1A2, CYP3A4 a P glykoproteinu
[5]. To vysvětluje mírné snížení expozice simvastatinu
prokázané v cílené klinické studii. V této studii
u 52 pacientů s NSCLC s prokázanou mutací EGFR,
průměrného věku 63,5 roku, z nichž pro srovnání farmakokinetiky simvastatinu bylo hodnotitelných 41, byl podáván
osimertinib v dávkách 80 mg jednou denně po dobu 30
dnů, před zahájením podávání osimertinibu a v 29. dni
jeho užívání byla podána jednorázová dávka simvastatinu
ve výši 40 mg [41]. Vlivem osimertinibu došlo ke snížení
AUC simvastatinu o 9 % (od zvýšení o 8 %
po snížení o 23 % při 90% CI) a ke snížení
jeho cmax o 23 % (6‒37 % při 90% CI). Inhibiční nebo indukční potenciál
inhibitorů EGFR shrnuje tabulka 9.
Lékové interakce erlotinibu se statiny
Erlotinib může být myotoxický. V odborné literatuře byla publikována kazuistická sdělení o vzniku rhabdomyolýzy při monoterapii erlotinibem [42] a při souběžném podávání erlotinibu s oxykodonem [43] nebo se simvastatinem [44]. Výrobce erlotinibu v ČR (v EU) uvádí [11]: „Kombinace erlotinibu se statiny může zvýšit možnost vzniku statinem indukované myopatie, včetně rhabdomyolýzy, která byla pozorována vzácně.“ Toto varování bylo začleněno do Souhrnu údajů o přípravku v roce 2010 na vyžádání Výboru pro humánní léčivé přípravky (Committee for Medicinal Products for Human Use, CHMP) Evropské lékové agentury (European Medicines Agency, EMA) po zhodnocení 6. pravidelně aktualizované zprávy o bezpečnosti (PSUR) (EMA: Tarceva Procedural Steps). Detaily nebyly zveřejněny a bez bližšího upřesnění bylo konstatováno, že nově identifikovaným rizikem je léková interakce erlotinibu se statiny (EMA: Tarceva: Variation assessment report, 7/2011 [45]). V dávkově eskalační studii s 18 pacienty (11 mužů/7 žen) [46] s pokročilými solidními zhoubnými nádory, ve věku 43‒70 let, byly u 5 z nich porovnány farmakokinetické vlastnosti erlotinibu podávaného v dávkách 150 mg jednou denně bez rosuvastatinu s farmakokinetickými vlastnostmi erlotinibu po 7 dnech souběžného podávání rosuvastatinu v dávkách 80 mg jednou denně (přibližně 1 mg/kg/den). Nebyly zjištěny statisticky významné změny farmakokinetických vlastností erlotinibu. Vliv erlotinibu na farmakokinetické vlastnosti rosuvastatinu nebylo možno vzhledem k uspořádání studie hodnotit (nebylo proveditelné podání samotného rosuvastatinu). Při dávkové eskalaci bylo hned při prvním zvýšení dávkování rosuvastatinu na 2 mg/kg denně (při souběžném podávání erlotinibu v dávkách 150 mg jednou denně) dosaženo dávku omezující toxicity, když se u 6 z 8 pacientů, jimž byla tato dávková úroveň podávána, vyskytly projevy myopatie, z toho v jednom případě došlo k rozvoji fatálně zakončené rhabdomyolýzy. Je ovšem třeba mít na paměti, že dávkování rosuvastatinu použitá v této studii představovala přibližně dvoj až čtyřnásobek maximální dávky rosuvastatinu v hypolipidemické indikaci.
Při terapii erlotinibem a souběžném podávání statinů je z výše uvedených důvodů třeba opatrnosti s ohledem na možné myotoxické nežádoucí účinky. Pacienty je třeba informovat o možném riziku myopatie a poučit je, aby v případě, že zaznamenají neobvyklou bolest, citlivost nebo slabost svalů, přestali rosuvastatin užívat a neprodleně vyhledali nebo přivolali lékařskou pomoc.
Lékové interakce inhibitorů EGFR v podmínkách běžné klinické praxe
V podmínkách ČR dosud nebyla
publikována žádná studie, která by se zabývala výskytem
lékových interakcí u pacientů léčených inhibitory
EGFR. Autoři proto vyjádřili očekávaný interakční potenciál
gefitinibu, erlotinibu, afatinibu a osimertinibu v podmínkách
běžné klinické praxe. Vycházeli přitom z údajů
o lékových interakcích 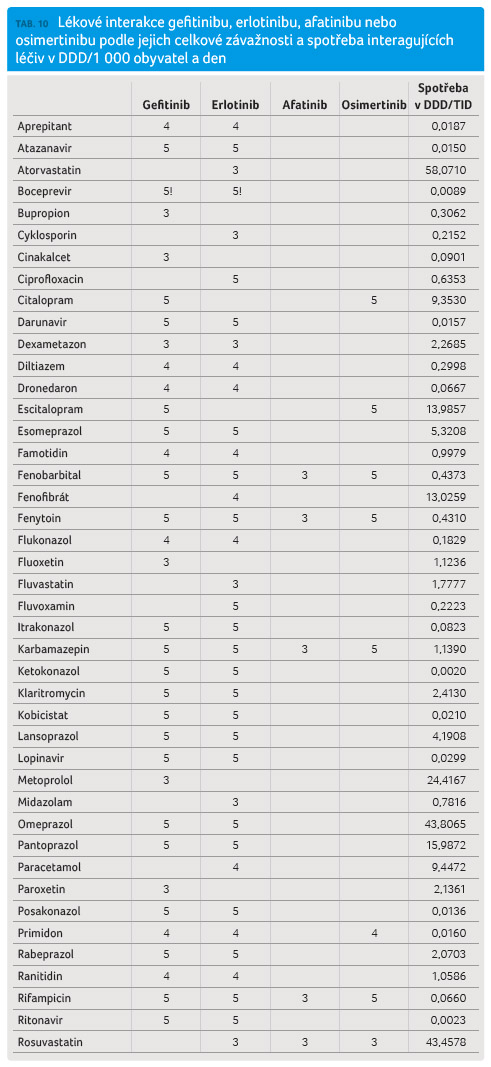 zmíněných léčiv publikovaných
v odborné literatuře nebo uvedených v oficiálních
dokumentech výrobců (a zveřejněných EMA nebo Úřadem pro
kontrolu potravin a léčiv, Food and Drug Administration, FDA),
přičemž v úvahu vzali pouze takové lékové interakce,
které mají podle Databáze lékových interakcí DrugAgency
závažnost stupně 3 a vyšší (dle zahraničních databází
lékových interakcí „moderate“ nebo „major“). Vedle toho
autoři zjistili na webových stránkách Státního ústavu pro
kontrolu léčiv (SÚKL) spotřebu interagujících
léčiv v DDD (definovaná denní dávka)/1 000 obyvatel
a den. Výsledky jsou uvedeny v tabulce 10.
zmíněných léčiv publikovaných
v odborné literatuře nebo uvedených v oficiálních
dokumentech výrobců (a zveřejněných EMA nebo Úřadem pro
kontrolu potravin a léčiv, Food and Drug Administration, FDA),
přičemž v úvahu vzali pouze takové lékové interakce,
které mají podle Databáze lékových interakcí DrugAgency
závažnost stupně 3 a vyšší (dle zahraničních databází
lékových interakcí „moderate“ nebo „major“). Vedle toho
autoři zjistili na webových stránkách Státního ústavu pro
kontrolu léčiv (SÚKL) spotřebu interagujících
léčiv v DDD (definovaná denní dávka)/1 000 obyvatel
a den. Výsledky jsou uvedeny v tabulce 10.
Nejvyšší četnost lékových
interakcí mají gefitinib a erlotinib, které vedou ke klinicky významným lékovým interakcím se silnými inhibitory a induktory
CYP3A4, protisekrečními léky a řadou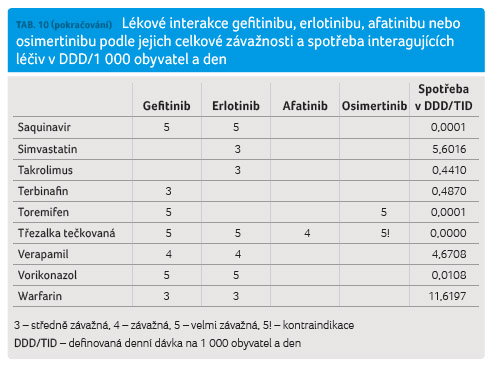 dalších léčiv.
Naproti tomu lékové interakce osimertinibu a zejména
afatinibu jsou podstatně méně četné a v případě afatinibu i méně klinicky významné.
dalších léčiv.
Naproti tomu lékové interakce osimertinibu a zejména
afatinibu jsou podstatně méně četné a v případě afatinibu i méně klinicky významné.
Vzhledem k tomu, že údaje uvedené v tabulce 10 neumožňují exaktní porovnání rizika lékových interakcí jednotlivých inhibitorů EGFR v podmínkách běžné klinické praxe, přistoupili autoři k dalším způsobům vyjádření tohoto rizika:
provedli součet spotřeby všech interagujících léčiv, čímž byla získána suma DDD/1 000 obyvatel a den vyjadřující počet osob z 1 000 obyvatel ČR, které každý den v roce užijí jednu definovanou denní dávku příslušného léčiva;
provedli
součin závažnosti lékové interakce se spotřebou příslušných
léčiv, čímž byly zís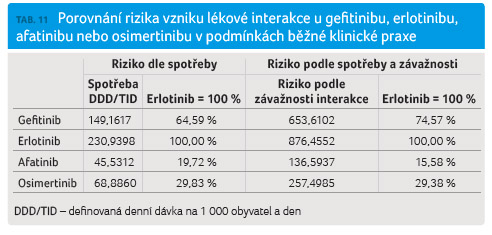 kány hodnoty zahrnující též závažnost
lékové interakce (avšak nevyjadřující vlastní riziko či
pravděpodobnost vzniku lékové interakce);
kány hodnoty zahrnující též závažnost
lékové interakce (avšak nevyjadřující vlastní riziko či
pravděpodobnost vzniku lékové interakce);
oba výše uvedené parametry pak byly upraveny na relativní vyjádření oproti erlotinibu, který má nejvyšší interakční potenciál ze všech zkoumaných léčiv.
Výsledky jsou shrnuty v tabulce 11.
Riziko vzniku lékové interakce v podmínkách běžné klinické praxe je nejvyšší v případě erlotinibu. Tento inhibitor EGFR má nejvíce lékových interakcí a tyto lékové interakce jsou klinicky relevantní. Nižší riziko vzniku lékových interakcí má gefitinib. Osimertinib má riziko vzniku lékových interakcí ve srovnání s erlotinibem méně než třetinové, a afatinib dokonce méně než pětinové.
Závěr
Při terapii lokálně pokročilého nebo metastazujícího NSCLC lze očekávat obdobný výskyt komorbidit jako v celé populaci obyvatel ČR. Tito pacienti budou patrně mít ve svém věku obvyklý výskyt arteriální hypertenze, dyslipidemie, hyperacidity, deprese a infekčních onemocnění (včetně mykotických chorob), alespoň se to ukazuje na populaci švédské [47]. V případě některých onemocnění lze jistě očekávat vyšší výskyt oproti věkově srovnatelné populaci (deprese, kognitivní deficit nebo infekční choroby) [48].
Z tohoto důvodu je podle názoru autorů možné použít údaje o spotřebě léčiv pro celou populaci ke kvantifikaci rizika vzniku lékové interakce u nemocných s NSCLC. Zcela jednoznačně vychází nejnižší riziko u léčiv, která mají nízký (osimertinib) nebo minimální (afatinib) interakční potenciál. Ten plyne z fyzikálně chemických vlastností těchto léčiv. Afatinib i osimertinib jsou dobře rozpustné ve vodě prakticky nezávisle na pH (za fyziologických podmínek), a proto tato léčiva na rozdíl od gefitinibu a erlotinibu neinteragují s protisekrečními léčivy. Současně afatinib i osimertinib buď nejsou substráty CYP3A4 (afatinib), nebo jsou jen málo citlivými substráty (osimertinib), resp. jsou málo citlivými substráty P glykoproteinu (obě léčiva), což je předpokladem pro malou klinickou závažnost příslušných lékových interakcí, kterých se lze v případě inhibitorů zcela vyvarovat vhodným načasováním podávání léčiv.
Cílem této práce nebylo hodnocení účinnosti jednotlivých terapeutických intervencí. Z literatury zabývající se lékovými interakcemi je však vcelku zřejmé, že se na účinnosti gefitinibu, erlotinibu, afatinibu a osimertinibu lékové interakce mohou poměrně významně podílet. Je známo, že současné podávání erlotinibu s protisekrečními léčivy může vést ke zhoršení prognózy nemocných. Je velmi pravděpodobné, že k podobnému výsledku dojde při terapii erlotinibem u kuřáků (pokud není dávka erlotinibu eskalována na dvojnásobek). Je též velmi pravděpodobné, že současné podávání zmíněných léčiv (patrně s výjimkou afatinibu) s induktory CYP3A4/P glykoproteinu povede též ke snížení účinku. Výrobci jednotlivých léčiv v ČR nabádají k monitorování účinnosti (což je pochopitelně možné např. sledováním farmakokinetických vlastností u konkrétního pacienta) nebo rovnou doporučují zvýšení dávky.
U pacienta s tuberkulózou bude třeba zvažovat podávání rifampicinu, neboť se jedná o silný induktor CYP3A4. Je však s podivem, že se nikdo z výrobců nezabývá středně silnými induktory CYP3A4 (efavirenz, bosentan, nafcilin, nevirapin, rifabutin, dexametazon, dabrafenib, oxkarbazepin, primidon) nebo slabými induktory CYP3A4 (aprepitant, modafinil, armodafinil, pioglitazon, prednison, metylprednison, eslikarbazepin, etravirin, rufinamid, topiramat, metamizol), případně celou plejádou léčivých rostlin (lékořice lysá, ženšen, šalvěj červenokořenná, rdesno hlavaté, jinan dvoulaločný, třapatka nachová, sója luštinatá, máta peprná a další), o nichž je vcelku věrohodně známo, že působí jako induktory CYP3A4. Kazuistika pacientky léčené gefitinibem a současně užívající kombinaci extraktu ze ženšenu a dřevokazných hub s cílem „zlepšit si imunitu“ je toho dobrou ilustrací. Je také zřejmé, že látky, které výše uvedené rostliny obsahují, z nichž některé se obchodují jako doplňky stravy (genistein jako izoflavon k mírnění potíží při menopauze), rovněž mají indukční vliv na CYP3A4. Genistein např. snížil plazmatické koncentrace jiného substrátu CYP3A4 midazolamu o 26 % [49]. Glycyrrhizin z lékořice lysé měl na midazolam podobný efekt [50], mentol měl též obdobný účinek na jiný substrát CYP3A4 ‒ triazolam [51]. Známá třezalka tečkovaná snižuje plazmatické koncentrace midazolamu o 53 % [52] a extrakt ze šalvěje červenokořenné dokonce o 58 % [53].
Mělo by být v zájmu zdravotních pojišťoven poskytnout svým klientům co nejširší informace. Nejde jen o finanční náročnost terapie, ale zejména o terapeutický úspěch a o co nejdelší prodloužení života jejich klientů.
Seznam použité literatury
- [1] Raju TN. The Nobel chronicles. 1986: Stanley Cohen Cohen and Rita Levi‑Montalcini. Lancet 2000; 355: 506.
- [2] Zhang Y, Zhang Z, Huang X, et al. Therapeutic efficacy comparison of 5 major EGFR‑TKIs in advanced EGFR‑positive non‑small‑cell lung cancer: a network meta‑analysis based on head‑to‑head trials. Clin Lung Cancer 2016; S1525–7304: 30231–30235.
- [3] Yang Z, Hackshaw A, Feng Q, et al. Comparison of gefitinib, erlotinib and afatinib in non‑small cell lung cancer: A meta‑analysis. Int J Cancer 2017; 140: 2805–2819.
- [4] Ding PN, Lord SJ, Gebski V, et al. Risk of treatment‑related toxicities from EGFR tyrosine kinase inhibitors: a meta‑analysis of clinical trials of gefitinib, erlotinib, and afatinib in advanced EGFR‑mutated non‑small cell lung cancer. J Thorac Oncol 2017; 12: 633–643.
- [5] Center for Drug Evaluation and Research. Application Number: 208065Orig1s000. Clinical Pharmacology and Biopharmaceutical Reviews of osimertinib, 2015. Dostupné na: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/208065Orig1s000ClinPharmR.pdf
- [6] Center for Drug Evaluation and Research. Application Number: 31‑399. Clinical Pharmacology and Biopharmaceutical Reviews of gefitinib, 2002. Dostupné na: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2003/21‑399_IRESSA_Clinr.pdf
- [7] Center for Drug Evaluation and Research. Application Number: 21–743. Clinical Pharmacology and Biopharmaceutical Reviews of erlotinib, 2004. Dostupné na: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2004/21–743_Tarceva_biopharmr.pdf
- [8] Center for Drug Evaluation and Research. Application Number: 201292Orig1s000. Clinical Pharmacology and Biopharmaceutical Reviews of afatinib, 2012. Dostupné na: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/201292Orig1s000ClinPharmR.pdf
- [9] https://en.wikipedia.org/wiki/Osimertinib
- [10] Iressa (gefitinib). Souhrn údajů o přípravku, 10/2016. Dostupné na: http://www.ema.europa.eu/docs/cs_CZ/document_library/EPAR_‑_Product_Information/human/001016/WC500036358.pdf
- [11] Tarceva (erlotinib). Souhrn údajů o přípravku, 4/2016. Dostupné na: http://www.ema.europa.eu/docs/cs_CZ/document_library/EPAR_‑_Product_Information/human/000618/WC500033994.pdf
- [12] Giotrif (afatinib). Souhrn údajů o přípravku, 11/2016. Dostupné na: http://www.ema.europa.eu/docs/cs_CZ/document_library/EPAR_‑_Product_Information/human/002280/WC500152392.pdf
- [13] Tagrisso (osimertinib). Souhrn údajů o přípravku, 1/2017. Dostupné na: http://www.ema.europa.eu/docs/cs_CZ/document_library/EPAR_‑_Product_Information/human/004124/WC500202022.pdf
- [14] Maher HM, Alzoman NZ, Shehata SM, Abahussain AO. UPLC‑ESI‑MS/MS study of the effect of green tea extract on the oral bioavailability of erlotinib and lapatinib in rats: Potential risk of pharmacokinetic interaction. J Chromatogr B Analyt Technol Biomed Life Sci 2017; 1049–1050: 30–40.
- [15] Hughes AN, O’Brien ME, Petty WJ, et al. Overcoming CYP1A1/1A2 mediated induction of metabolism by escalating erlotinib dose in current smokers. J Clin Oncol 2009; 27: 1220–1226.
- [16] Swaisland HC, Ranson M, Smith RP, et al. Pharmacokinetic drug interactions of gefitinib with rifampicin, itraconazole and metoprolol. Clin Pharmacokinet 2005; 44: 1067–1081.
- [17] Rakhit A, Pantze MP, Fettner S, et al. The effects of CYP3A4 inhibition on erlotinib pharmacokinetics: computer‑based simulation (SimCYP) predicts in vivo metabolic inhibition. Eur J Clin Pharmacol 2008; 64: 31–41.
- [18] Hamilton M, Wolf JL, Drolet DW, et al. The effect of rifampicin, a prototypical CYP3A4 inducer, on erlotinib pharmacokinetics in healthy subjects. Cancer Chemother Pharmacol 2014; 73: 613–621.
- [19] Wind S, Giessmann T, Jungnik A, et al. Pharmacokinetic drug interactions of afatinib with rifampicin and ritonavir. Clin Drug Investig 2014; 34: 173–182.
- [20] NCT02157883. Dostupné na: http://clinicaltrials.gov/show/NCT02157883
- [21] FDA Full Prescription Information: Iressa (gefitinib), AstraZeneca, 7/2015. Dostupné na: http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/206995s000lbl.pdf
- [22] FDA Full Prescription Information: Tarceva (erlotinib), Genetech, 10/2016. Dostupné na: http://www.accessdata.fda.gov/drugsatfda_docs/label/2016/021743s025lbl.pdf
- [23] FDA Full Prescription Information: Giotrif (afatinib), Boehringer Ingelheim, 4/2016. Dostupné na: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/201292s010lbl.pdf
- [24] FDA Full Prescription Information: Tagrisso (osimertinib), AstraZeneca, 3/2017. Dostupné na: http://www.accessdata.fda.gov/drugsatfda_docs/label/2017/208065s006lbl.pdf
- [25] Hwang SW, Han HS, Lim KY, Han JY. Drug interaction between complementary herbal medicines and gefitinib. J Thorac Oncol 2008; 3: 942–943.
- [26] Kletzl H, Zwanziger E, Kirkpatrick C, Luedin E. Effect of ciprofloxacin on the systemic exposure to erlotinib. J Clin Oncol 2008; 26 (Suppl 15): 19047–19047.
- [27] Gass‑Jégu F, Gschwend A, Gairard‑Dory AC, et al. Gastrointestinal perforations in patients treated with erlotinib: A report of two cases with fatal outcome and literature review. Lung Cancer 2016; 99: 76–78.
- [28] Dobrinas M, Cornuz J, Oneda B, et al. Impact of smoking, smoking cessation, and genetic polymorphisms on CYP1A2 activity and inducibility. Clin Pharmacol Ther 2011; 90: 117–125.
- [29] Anderson GD, Chan LN. Pharmacokinetic drug interactions with tobacco, cannabinoids and smoking cessation products. Clin Pharmacokinet 2016; 55: 1353–1368.
- [30] Kobayashi H, Sato K, Niioka T, et al. Effects of polymorphisms in CYP2D6 and ABC transporters and side effects induced by gefitinib on the pharmacokinetics of the gefitinib metabolite, O‑desmethyl gefitinib. Med Oncol 2016; 33: 57.
- [31] Suzumura T, Kimura T, Kudoh S, et al. Reduced CYP2D6 function is associated with gefitinib‑induced rash in patients with non‑small cell lung cancer. BMC Cancer 2012; 12: 568.
- [32] Takimoto T, Kijima T, Otani Y, et al. Polymorphisms of CYP2D6 gene and gefitinib‑induced hepatotoxicity. Clin Lung Cancer 2013; 14: 502–507.
- [33] van Leeuwen RWF, Jansman FGA, Hunfeld NG, et al. Tyrosine kinase inhibitors and proton pump inhibitors: an evaluation of treatment options. Clin Pharmacokinet 2017; 56: 683–688.
- [34] Tang W, Tomkinson H, Masson E. Effect of sustained elevated gastric pH levels on gefitinib exposure. Clin Pharmacol Drug Dev 2017. doi: 10.1002/cpdd.337 [in press]
- [35] Kletzl H, Giraudon M, Ducray PS, et al. Effect of gastric pH on erlotinib pharmacokinetics in healthy individuals: omeprazole and ranitidine. Anticancer Drugs 2015; 26: 565–572.
- [36] Chu MP, Ghosh S, Chambers CR, et al. Gastric acid suppression is associated with decreased erlotinib efficacy in non‑small‑cell lung cancer. Clin Lung Cancer 2015; 16: 33–39.
- [37] van Leeuwen RW, Peric R, Hussaarts KG, et al. Influence of the acidic beverage cola on the absorption of erlotinib in patients with non‑small‑cell lung cancer. J Clin Oncol 2016; 34: 1309–1314.
- [38] NCT02224053. Dostupné na: http://clinicaltrials.gov/ct2/show/NCT02224053
- [39] Liu Y, Ramírez J, House L, Ratain MJ. The UGT1A1*28 polymorphism correlates with erlotinib’s effect on SN‑38 glucuronidation. Eur J Cancer 2010; 46: 2097–2103.
- [40] NCT02317016. Dostupné na: http://clinicaltrials.gov/show/NCT02317016
- [41] NCT02197234. Dostupné na: http://clinicaltrials.gov/show/NCT02197234
- [42] Moscetti L, Nelli F, Ruggeri EM. Rhabdomyolysis from erlotinib: a case report. Tumori 2011; 97: 415–416.
- [43] Koršić M, Muršić D, Badovinac S, et al. Erlotinib‑related rhabdomyolysis: the role of pharmacogenetics and drug‑drug interaction. Cancer Chemother Pharmacol 2015; 76: 1317–1399.
- [44] Veeraputhiran M, Sundermeyer M. Rhabdomyolysis resulting from pharmacologic interaction between erlotinib and simvastatin. Clin Lung Cancer 2008; 9: 232–234.
- [45] Goss GD, Jonker DJ, Laurie SA, et al. A phase I study of high‑dose rosuvastatin with standard dose erlotinib in patients with advanced solid malignancies. J Transl Med 2016; 14: 83.
- [46] EMA: Tarceva. Variation assessment report, 7/2011. Dostupné na: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‑_Assessment_Report_‑_Variation/human/000618/WC500117593.pdf
- [47] Nilsson J, Berglund A, Bergström S, et al. The role of comorbidity in the management and prognosis in non‑small cell lung cancer: a population‑based study. Acta Oncol 2017; 56: 949–956.
- [48] Schulkes KJ, Hamaker ME, van den Bos F, van Elden LJ. Relevance of a geriatric assessment for elderly patients with lung cancer – a systematic review. Clin Lung Cancer 2016; 17: 341–349.
- [49] Xiao CQ, Chen R, Lin J, et al. Effect of genistein on the activities of cytochrome P450 3A and P‑glycoprotein in Chinese healthy participants. Xenobiotica 2012; 42: 173–178.
- [50] Tu JH, He YJ, Chen Y, et al. Effect of glycyrrhizin on the activity of CYP3A enzyme in humans. Eur J Clin Pharmacol 2010; 66: 805–810.
- [51] Hoshino M, Ikarashi N, Hirobe R, et al. Effects of menthol on the pharmacokinetics of triazolam and phenytoin. Biol Pharm Bull 2015; 38: 454–460.
- [52] Hall SD, Wang Z, Huang SM, et al. The interaction between St John’s wort and an oral contraceptive. Clin Pharmacol Ther 2003; 74: 525–535.
- [53] Qiu F, Jiang J, Ma Y, et al. Opposite effects of single‑dose and multidose administration of the ethanol extract of danshen on CYP3A in healthy volunteers. Evid Based Complement Alternat Med 2013; 2013: 730–734.