Lékové interakce antidepresiv s pohledem do klinické praxe
Souhrn:
Tašková I. Lékové interakce antidepresiv s pohledem do klinické praxe. Remedia 2022; 32: 74–80.
Téměř každé léčivo může interagovat s dalšími léčivy, doplňky stravy, potravinami nebo návykovými látkami. Léková interakce ve smyslu léčivo–léčivo nastává, pokud jeden lék mění účinky druhého. Výsledkem jejich vzájemného působení je většinou snížení nebo zvýšení účinku. Pacient je v prvním případě ohrožen neúčinností terapie, ve druhém případě zase vystupňovanými nežádoucími účinky, které mohou vést k hospitalizaci nebo k jejímu prodloužení, což následně vede k vyšší ekonomické zátěži zdravotnického systému. Antidepresiva jsou léky, které pacienti užívají často dlouhodobě, mnohdy celoživotně. Hlubší znalosti v oblasti jejich lékových interakcí mohou vést ke zvýšení bezpečnosti lékových režimů našich pacientů a současně k ekonomickým úsporám. Pro správnou interpretaci klinické významnosti lékových interakcí jsou potřebné solidní znalosti farmakokinetiky (zejména informace o způsobu metabolizace léčiva) i farmakodynamiky léčiv (oblast mechanismu účinku a rizik, jež léčivo přináší). Optimální je mít také základní přehled o silných induktorech a inhibitorech cytochromu P450 (hlavních enzymů první fáze biotransformace) a také mít povědomí o tom, které jeho izoformy podléhají genetickému polymorfismu. Tento článek přináší základní teoretický přehled problematiky lékových interakcí antidepresiv. Ten je navíc doplněn o pohled z praxe klinického farmaceuta, a to formou dvou kazuistik, jejichž ústředním tématem jsou právě lékové interakce antidepresiv.
Summary:
Taskova I. Drug‑drug interactions of antidepressants and the case studies. Remedia 2022; 32: 74–80.
Almost any drug can interact with other medications, dietary supplements, foods, or illicit substances when administered to a patient. A drug interaction occurs when one drug modifies the effects of another drug. The result is usually a reduction or enhancement of their effect. In the first case, the patient is endangered by the ineffectiveness of the therapy, in the second case by exacerbated side effects, which may lead to hospitalisation or prolongation of hospitalisation stay, leading to a higher economic burden on the health care system. Antidepressants are drugs that patients often take for a long time. Profound knowledge of drug interactions can lead to increased safety of our patients' drug regimens and, at the same time, to economic savings. To correctly interpret the clinical significance of drug interaction, a solid knowledge of pharmacokinetics and pharmacodynamics of the drug is required. The theoretical overview of the antidepressants drug interactions topic, which is presented in this article, is supplemented by a practical point of view of a clinical pharmacist in the form of two case reports.
Key words: drug interactions, antidepressants, SSRI, serotonin syndrome
Úvod
Téměř každé léčivo může interagovat s dalšími léčivy, doplňky stravy, potravinami nebo návykovými látkami. Léková interakce ve smyslu léčivo–léčivo nastává, pokud jeden lék mění účinky druhého. Výsledkem jejich vzájemného působení je pak snížení nebo zvýšení jejich účinku. Pacient je v prvním případě ohrožen neúčinností terapie, ve druhém případě zase vystupňovanou toxicitou léčiva.
Lékové interakce zvyšují morbiditu i mortalitu pacientů [1]. Přítomnost lékové interakce vyvolává až ve čtvrtině případů projevy nežádoucích účinků, které mohou také vést k hospitalizaci nebo k prodloužení její délky (o 1,75, resp. 2,9 dne), což představuje vyšší ekonomickou zátěž zdravotnického systému [2,3]. Dalším následkem projevu nežádoucího účinku může být snížená adherence pacienta k terapii, což je také spojováno s vyšší mírou hospitalizace [1,4].
Dechanontová se svým thajským kolektivem autorů uskutečnila v roce 2014 rozsáhlou metaanalýzu dat pocházejících zejména ze západních zemí (Evropa, USA) a konkrétně se snažila popsat souvislost mezi přijetím pacienta k hospitalizaci a přítomností klinicky závažné interakce v lékovém režimu. Ze studie vyplynulo, že 1,1 % pacientů je hospitalizováno právě z důvodu takové lékové interakce [4]. Metaanalýza, kterou provedl brazilský kolektiv Gonzagové v roce 2020 na souboru přibližně 6 500 již hospitalizovaných pacientů, dospěla k závěru, že klinicky závažnou interakcí je ohroženo asi 10 % hospitalizovaných pacientů [5]. Je tedy jasné, že hlubší znalosti v oblasti lékových interakcí by mohly vést ke zvýšení bezpečnosti lékových režimů našich pacientů a současně k ekonomickým úsporám v podobě snížené míry hospitalizace či zkrácení její délky.
Typy lékových interakcí
Existují dva základní typy lékových interakcí. Farmakodynamická interakce je výsledkem působení obou látek na cílovém místě (např. receptor, enzym apod.), na kterém mohou obě látky účinkovat synergicky (účinek se sčítá), nebo antagonisticky (účinek je protichůdný). Naopak farmakokinetické interakce vznikají mimo cílová místa léčiv, a to během farmakokinetických dějů (absorpce, distribuce, metabolizace a exkrece) a výsledkem jsou změny v plazmatické koncentraci léčiv, a tudíž změna jejich dostupnosti na cílovém místě [1,6].
V popředí zájmu každého lékaře a farmaceuta by měly být ty interakce, které jsou klinicky významné. Znalost lékových interakcí antidepresiv je jedním ze základních předpokladů správné a bezpečné terapie v psychiatrii.
V praxi můžeme vidět dva extrémy. V jednom případě panuje obava z interakcí až do té míry, že mnohdy brání nasazení účinné terapie, která může být za podmínek monitorace rizika zcela bezpečná. Opačným extrémem jsou případy souběžné preskripce léčiv, jež spolu interagují do takové míry, že se efekt jednoho z nich nemůže projevit, nebo se naopak již projevuje nežádoucími účinky, v krajním případě až intoxikací. Ani jedna z výše popsaných situací by neměla nastat a jedinou vhodnou prevencí jejího vzniku je správná interpretace lékových interakcí na základě znalosti interakčního potenciálu jednotlivých látek včetně znalosti rizik, která jednotlivá léčiva přinášejí.
Farmakokinetika antidepresiv
Většina antidepresiv jsou malé
lipofilní molekuly. Jedná se tedy o látky s dobrou
absorpcí z gastrointestinálního traktu (GIT) a s dobrou
biologickou dostupností, se snadným přestupem přes
hematoencefalickou bariéru, s velkým distribučním objemem
(Vd), s lineární farmakokinetikou a velká část z nich
je metabolizována játry zpravidla na neaktivní
metabolity (nejčastěji za pomoci cytochromu P450). Existují
ale i výjimky (např. delší/kratší biologický poločas,
existence aktivního metabolitu, významná renální eliminace
apod.), které je potřeba znát, abychom lékové interakce dokázali
správně interpretovat (více viz tab. 1) [7,8].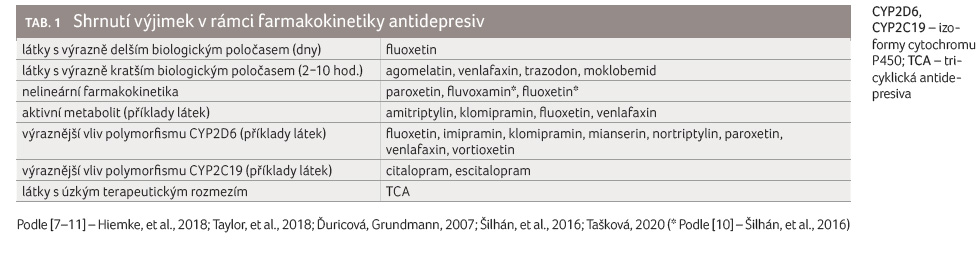
Interakce antidepresiv
Farmakodynamické interakce
Farmakodynamické interakce jsou méně častým typem interakcí a vycházejí z receptorového profilu, tj. z mechanismu účinku.
Antidepresiva jsou vesměs serotonergní
a noradrenergní, v jednom případě i dopaminergní
(bupropion) léčiva. Mohou tedy interagovat na receptorech
5 HT, α a D. Látky, které inhibují zpětné vychytávání
serotoninu a noradrenalinu (případně i dopaminu), pak
mohou interagovat i na transportérech těchto
neurotransmiterů (SERT – membránový přenašeč pro
serotonin, NET – membránový přenašeč pro
noradrenalin, DAT – membránový přenašeč pro
dopamin). Některé látky také vykazují anticholinergní,
antihistaminergní a antiadrenergní
nežádoucí účinky. I na těchto receptorech (M1,
H1, α1 a α2) mohou vznikat
farmakodynamické lékové interakce, stejně jako na některých
iontových kanálech (sodíkové, draslíkové), jež některá
z antidepresiv také ovlivňují.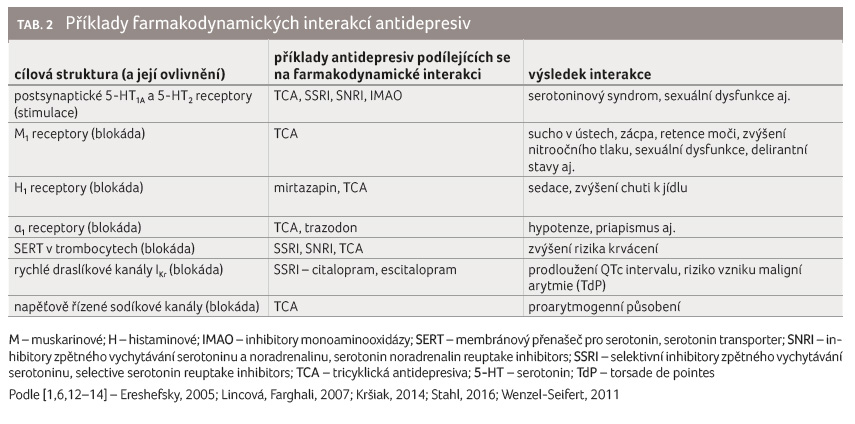
Příklady nejčastějších farmakodynamických interakcí antidepresiv jsou uvedeny v tabulce 2.
Farmakokinetické interakce
Jak již bylo zmíněno, farmakokinetické interakce jsou druhem interakcí, kdy se mění dostupnost léčiva na cílovém místě díky změnám v absorpci, distribuci, metabolismu nebo exkreci vlivem dalšího léčiva.
Ke změně absorpce dojde například při podání kolesevelamu (pryskyřice, hypolipidemikum), který může snížit biodostupnost všech současně perorálně podaných léčiv.
Příkladem interakce se změnou distribuce může být vytěsnění warfarinu z vazby na albumin molekulou selektivního inhibitoru zpětného vychytávání serotoninu (selective serotonin reuptake inhibitor, SSRI) s vysokou vazbou na tuto plazmatickou bílkovinu (fluoxetin, sertralin, paroxetin), což může mít za následek zvýšení hodnoty mezinárodního normalizovaného poměru (international normalized ratio, INR) nad terapeutické rozmezí. Tento druh interakce nebývá však klinicky významný, pokud má pacient funkční eliminační orgány a podstupuje pravidelný monitoring INR. Následně po vytěsnění z vazby totiž dochází k ustálení nového rovnovážného stavu mezi volnou a vázanou frakcí a volná frakce je poté eliminována. Příkladem interakce na úrovni exkrece, které také nemívají v případě antidepresiv zásadní význam, může být kompetice o transportéry v ledvinných tubulech nebo změna exkrece léčiva z důvodu změny pH moči [6,15,16].
Nejčastěji za farmakokinetickými interakcemi stojí změny metabolismu léčiv. Tyto interakce mohou být klinicky významné.
Léčiva obecně jsou poměrně lipofilní látky. Aby mohla být vyloučena z organismu renálně, je potřeba je upravit do hydrofilnější podoby. Proto vznikl během fylogenetického vývoje člověka velmi důmyslný systém intracelulárních metabolických enzymů lokalizovaných zejména v hepatocytech, které ve dvou fázích upravují léčiva do podoby, v níž mohou být z těla vyloučena, a většinou u toho ztrácejí i svou biologickou aktivitu. První fáze biotransformace léčiv zpravidla probíhá na cytochromu P450 (CYP). Jeho izoformy jsou substrátově specifické a probíhají na nich oxidačně redukční reakce (např. hydroxylace). Léčivo získává nové hydrofilní skupiny, které mění jeho fyzikálně chemické vlastnosti, tedy i rozpustnost ve vodě. Většina léčiv se biotransformuje pomocí izoforem CYP3A4 (30 %), CYP2D6 (20 %), CYP2C9 (13 %), CYP1A2 (9 %), CYP2B6 (7 %) a CYP2C19 (7 %). Druhá fáze biotransformace léčiv spočívá v různých konjugačních reakcích (např. glukuronidace), což znamená, že na léčivo upravené v první fázi se váže velká organická molekula (např. kyselina glukuronová). Vznikem konjugátu se ještě zvýší hydrofilita léčiva. Ten je následně z organismu eliminován. Některá léčiva jsou svou povahou dostatečně hydrofilní, proto jejich metabolizace probíhá rovnou druhou fází biotransformace bez účasti CYP (např. valproát, oxazepam) [16–19].
Plazmatickou koncentraci léčiva může
významně snížit interakce s induktorem cytochromu P450,
který tak urychlí jeho eliminaci z organismu. To může
ve výsledku vést k nižší účinnosti léčiva, protože
je rychleji metabolizováno větším množstvím enzymu. Nástup
této interakce je obyčejně opožděn (7–10 dní) od nasazení
induktoru, jelikož je potřeba, aby se enzym v organismu
nasyntetizoval. Interakce přetrvává zhruba stejnou dobu
po vysazení induktoru (7–10 dní), kdy je potřeba vyčkat
eliminace induktoru z organismu (5 biologických poločasů
induktoru) a dále degradace enzymu (5 biologických poločasů
enzymu, tj. zhruba týden). Enzymatický inhibitor naopak eliminaci
zpomalí, což může vést ke kumulaci léčiva v organismu
a následně k projevům nežádoucích účinků či
toxicity. Nástup inhibice po nasazení inhibitoru je téměř
okamžitý a jeho vliv pomine opět po eliminaci inhibitoru
z organismu (5 biologických poločasů inhibitoru).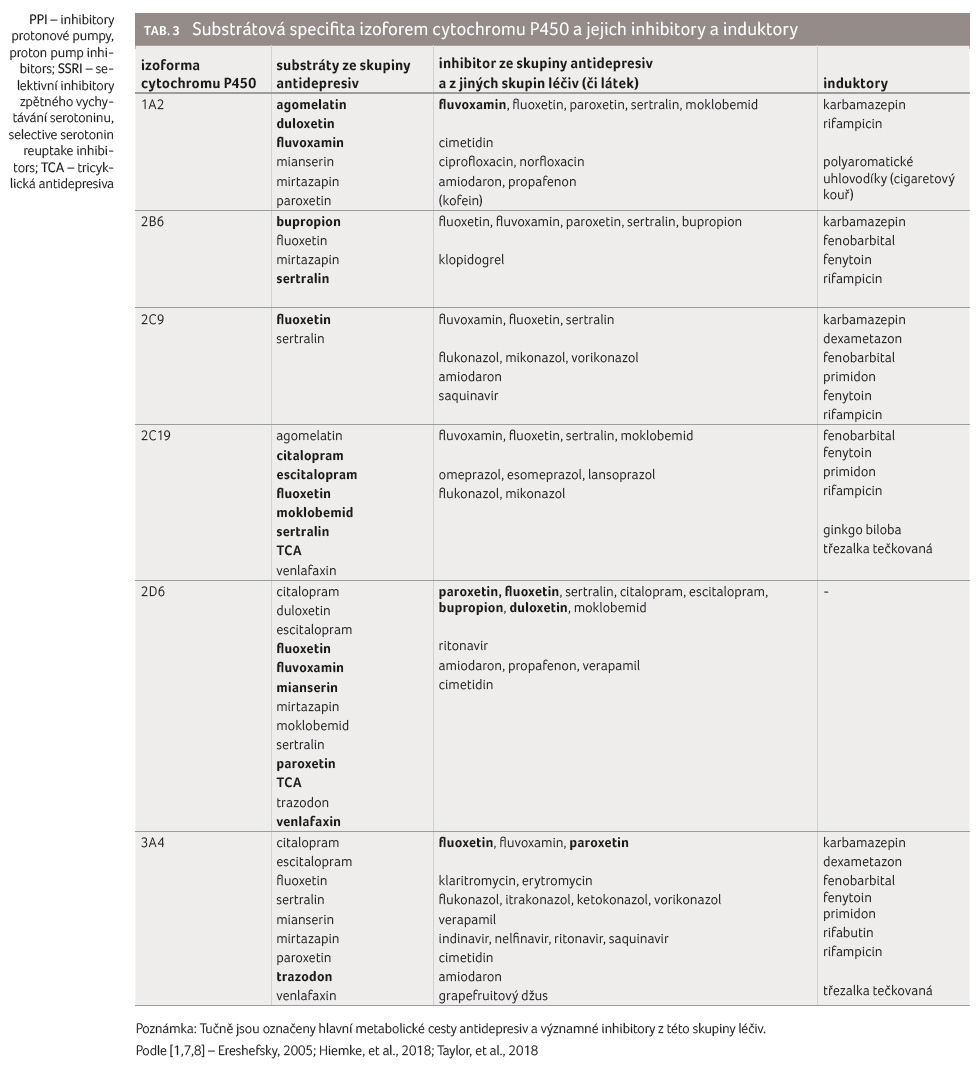
Přehled substrátové specifity hlavních izoforem cytochromu P450 a jejich hlavních inhibitorů a induktorů z řad antidepresiv a případně i jiných skupin léčiv přehledně uvádí tabulka 3.
Další oblast, jejíž znalost je podstatná pro správnou interpretaci lékových interakcí, představuje genetický polymorfismus izoforem cytochromu P450 CYP2D6, CYP2C19 a CYP2C9. Jedinci v populaci mohou mít tedy různě funkční alely (varianty genu kódující danou izoformu). Výsledkem je velká interindividuální variabilita v aktivitě těchto izoforem, na jejímž základě lze rozdělit populaci na pomalé (defektní gen), intermediární (jedna defektní a jedna intaktní alela), rychlé (intaktní alely) a ultrarychlé (duplikace nebo amplifikace genomu) metabolizátory. Pomalí metabolizátoři jsou vystaveni vyššímu riziku toxicity, pokud užívají léčiva, která se primárně metabolizují touto cestou, neboť následně dochází ke kumulaci mateřské látky. Ultrarychlý metabolizátor je naopak ohrožen neúčinností terapie z důvodu rychlejší metabolizace a následné eliminace léčiva.
Pokud je v lékovém režimu přítomna léková interakce ovlivňující aktivitu výše zmíněných izoforem CYP, je potřeba brát tuto skutečnost jako důležitý faktor, který může zásadně ovlivnit výsledek interakce. Ilustrativním příkladem může být pacient, který užívá kombinaci antidepresiv bupropion a venlafaxin. Předpokladem je, že bupropion (silný inhibitor CYP2D6) bude zvyšovat plazmatické koncentrace venlafaxinu (substrátu CYP2D6) několikanásobně. Pokud však pacient bude ultrarychlým metabolizátorem, výsledek interakce se neprojeví. Naopak, pokud by pacient byl např. pomalým nebo intermediárním metabolizátorem, ocitne se ve vysokém riziku intoxikace venlafaxinem. Problematické je, že v běžném provozu nemocnice nejsme schopni zjistit, kterým metabolizátorem náš pacient je [7,9,16,20]. Substráty izoforem, jež vykazují genetický polymorfismus (CYP2D6, CYP2C9 a CYP2C19) z řad antidepresiv, jsou uvedeny v tabulce 3.
Dalším faktorem, který může také
výrazně ovlivnit absorpci, distribuci a eliminaci řady léčiv,
je aktivita efluxního transmembránového transportéru
P glykoproteinu (P gp), který je důležitým
obranným mechanismem před vstupem xenobiotik do organismu.
Tento přenašeč je strategicky situován v epiteliálních
buňkách orgánů podílejících se na absorpci a distribuci
látek, jako jsou enterocyty, buňky hematoencefalické, testikulární
a placentární bariéry, a také v lokalizacích
s čistě exkreční funkcí – v kanalikulární
membráně hepatocytů, na apikální straně epitelových buněk
žlučových kanálků a renálního proximálního tubulu.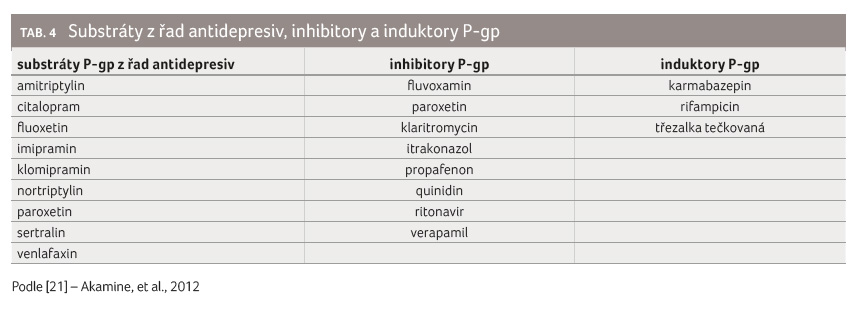
Pro P gp je typické, v porovnání s ostatními transportéry, že s ním interaguje velké množství látek, často chemicky i funkčně značně odlišných. Mezi jeho substráty patří nejen léčiva z různých farmakoterapeutických skupin (např. digoxin, amiodaron, inhibitory HIV proteáz, antimykotika, antibiotika, celá řada chemoterapeutik a imunosupresiv), ale i endogenní hormony a jiné látky (např. cholesterol, kortizol). Dle některých zdrojů patří mezi substráty P gp i některá antidepresiva (viz tab. 4). Podobně jako cytochrom P450 může mít i P gp své induktory a inhibitory, které mohou pak snižovat či zvyšovat plazmatickou koncentraci substrátu. Mnoho substrátů, induktorů i inhibitorů P gp má velký překryv se substráty, induktory a inhibitory cytochromu P450 (zejména s izoformou CYP3A4).
V genu MDR1, který kóduje protein P gp, je také popisován vysoký výskyt bodových polymorfismů. Nadměrná exprese genu MDR1 často vede k rezistenci na podávanou terapii. Například u nádorových buněk je považována za jednu z významných příčin rozvoje mnohočetné lékové rezistence (multidrug resistance, MDR); ten se z důvodu široké substrátové specifity týká i léčiv, kterým nádorové buňky nebyly ještě nikdy vystaveny (teoreticky i výše vyjmenovaných antidepresiv). Přítomnost polymorfismů v genu MDR1 je také často studována ve spojitosti s výskytem celé řady onemocnění (akutní lymfoblastická leukemie, kolorektální karcinom, farmakorezistentní epilepsie a jiné) [16,21,22].
Co nám v interpretaci lékových
interakcí může v praxi pomoci?
Při interpretaci lékových interakcí je v první řadě nutná hlubší znalost farmakodynamiky a farmakokinetiky léčiv. Vhodné je se zabývat zejména těmito otázkami:
- Jakými mechanismy léčivo působí?
- Co je cílovým místem léčiva?
- Jedná se o léčivo s úzkým terapeutickým indexem?
- Jaké má nežádoucí účinky a jsou na dávce závislé, či nezávislé?
- Jakými způsoby je léčivo metabolizováno a eliminováno (jedna, nebo více paralelních metabolických cest, existence aktivních metabolitů aj.)?
- V jaké formě je léčivo podáváno (forma proléčiva, nebo aktivní látka)?
- Která léčiva mohou jeho farmakokinetiku ovlivňovat a naopak, u kterých léčiv může dané léčivo farmakokinetiku ovlivňovat?
- Může ovlivnit metabolismus léčiva genetický polymorfismus některého z enzymů/přenašečů?
- Které další doplňky stravy, potraviny nebo návyky (pití kávy, kouření) mohou účinnost léčiv ovlivnit?
- Je možné monitorovat jeho plazmatickou koncentraci a je toto vyšetření dostupné?
Mnoho kliniků spoléhá na různé typy interakčních softwarů, které mohou být do jisté míry užitečným nástrojem k identifikaci lékových interakcí. Úskalí těchto programů je však několik. Jednak interakční software popisuje závažnost a výsledek interakce na základě populačního průměru. Náš pacient však nemusí být tímto průměrným pacientem a může reagovat na podání interagujících léčiv odlišně, a proto nelze na interakční software spoléhat absolutně a učinit závěr o interakci čistě z upozornění na její existenci. Software za nás není schopen interakci interpretovat v kontextu pacientova zdravotního stavu, komorbidit a dalších rizikových faktorů. To může jen klinik, který je s pacientským případem obeznámen a má k dispozici jeho kompletní dokumentaci. Smyslem interakčních softwarů může být poskytnutí možnosti uhlídat interakční potenciál komplikovaných lékových režimů u polymorbidních pacientů. Lze ho využít jako jakýsi základní soubor informací, ze kterého pak můžeme díky klinické zkušenosti a znalosti dat o pacientovi učinit závěr o klinické závažnosti interakce a zareagovat úpravou dávky či záměnou léčiva, pokud je to nutné.
Nadměrná důvěra v interakční software vede často k situacím, kdy se pacientovi nepodá lék, který je u něj indikován, ze strachu z interakce, kterou například můžeme ošetřit monitorováním rizik nebo je její klinický význam malý. Pacient tak může být ochuzen o bezpečné a účinné léčivo, namísto kterého se pak podávají méně účinná a více riziková léčiva, nebo dokonce jejich kombinace. Druhou chybou nebo extrémem je, pokud klinik přítomnost interakcí bagatelizuje, nezamýšlí se nad nimi nebo jejich projevy přičítá onemocnění či pouze nežádoucímu účinku léčiv. Pacient pak může zbytečně zažívat nepříjemné vedlejší projevy léčby (viz kazuistiku 1).
K objektivizaci či interpretaci lékové interakce u konkrétního pacienta lze použít metodu terapeutického monitorování plazmatických koncentrací léčiv (TDM). TDM využívá stanovení koncentrace léčiva v krevní plazmě nebo séru k dávkové optimalizaci tak, aby bylo dosaženo maximální účinnosti léku s co nejnižším rizikem toxicity. Je prokázáno, že mezi pacienty existuje při podání stejné dávky velká interindividuální variabilita ve farmakokinetice léčiv, některé zdroje udávají až dvacetinásobný rozdíl v plazmatické koncentraci jejich ustálených lékových koncentrací (Css) [7,10]. Pokud máme podezření na lékovou interakci např. při výrazně vyjádřeném nežádoucím účinku léku při jeho běžném dávkování nebo naopak při jeho neúčinnosti i přes prokázanou compliance pacienta, lze ověřit naši hypotézu stanovením plazmatické koncentrace daného léku, pokud je toto vyšetření u daného léku k dispozici.
Závěrem lze shrnout postup zhodnocení relevance lékové interakce např. následovně:
- znalost kontraindikovaných a klinicky závažných interakcí psychofarmak;
- znalost rizikových faktorů pro vyšší citlivost na lékové interakce na straně pacienta (polymorbidita, vyšší věk, změněná funkce eliminačních orgánů, genetické vlivy, změny v homeostáze aj.);
- posouzení rizik léčiva pro daného pacienta;
- posouzení možnosti monitorace těchto rizik (laboratoř, fyzikální vyšetření nebo vyšetření specialistou);
- posouzení existence jiné možnosti terapie;
- vyhledání dat, která interakci popisují;
- využití terapeutického monitorování léčiv, pokud je vyšetření racionální a dostupné;
- zvážení jiné alternativy léčby nebo úpravy dávky, pokud je interakce závažná nebo se u pacienta objevily nežádoucí projevy léčby, které by mohly být výsledkem interakce;
- dokumentace klinicky významné interakce.
Tématu interakcí je potřeba věnovat dostatečnou pozornost, jelikož antidepresiva pacienti užívají mnohdy dlouhodobě či celoživotně. Antidepresiva mohou ovlivňovat efektivitu léčiv, která pacienti užívají souběžně, a naopak tato léčiva mohou ovlivňovat účinnost, snášenlivost a v krajním případě i bezpečnost antidepresivní léčby. Pro ilustraci lékových interakcí antidepresiv a jejich řešení jsou uvedeny závěrem následující pacientské případy.
Kazuistika 1: Polékový třes jako projev interakce trazodonu a fluoxetinu
Základní informace:
Žena, 21 let
Dg.: úzkostně depresivní syndrom, hraniční porucha osobnosti a porucha příjmu potravy
Epikríza:
Pacientka byla hospitalizována (4. hospitalizace) po delší době sebepoškozování, krátce po TS (pokus o sebevraždu, tentamen suicidii) škrcením, který po přijetí hodnotila jako impulzivní. Po základní stabilizaci byla přeložena na psychoterapeutické oddělení s plánem další stabilizace a terapeutické práce na záchvatovitém přejídání a impulzivním chování.
Medikace při přijetí:
fluoxetin 20 mg tbl. 2 0 0
trazodon 300 mg tbl. 1 tbl. na noc
s prodlouženým uvolňováním
oxazepam 20 mg tbl. 1 tbl. při potížích
(max. 4×/den, min. interval
podání 3 hod.)
Poznámka: Medikace byla ponechána, pacientce vyhovuje.
Objektivní nález: Somaticky je zdravá, pouze poslední rok popisuje obtěžující tremor horní končetiny, který přetrvá celý den, zhoršuje se při stresu a do jisté míry již brání sebeobsluze. Pro přetrvávající tremor je neurologicky vyšetřena se závěrem v.s. incipientní familiární esenciální tremor s doporučením prozatím nemedikovat, kontrola d.p.
Lékař poté oslovuje oddělení klinické farmacie pro konzultaci možné polékové příčiny tremoru, který je pro pacientku velmi obtěžující.
Konzilium klinického farmaceuta:
Po vyloučení jiných příčin třesu lze zvažovat jako možnou příčinu přesycení pacientky serotoninem v důsledku lékové interakce dvou serotonergních léčiv – fluoxetinu (silného inhibitoru CYP2D6 a CYP3A4) a trazodonu (substrátu CYP2D6 a CYP3A4). Dávám ke zvážení odběr plazmatické koncentrace trazodonu (v.s. intoxikace trazodonem).
Další vývoj:
Pacientce byla odebrána pro v.s. intoxikaci trazodonem jeho plazmatická koncentrace a následující den byla jeho dávka snížena na polovinu (tj. na 150 mg/den), za 5 dní na čtvrtinu (75 mg/den) a za dalších 5 dní byl trazodon vysazen zcela. Fluoxetin byl v medikaci ponechán jako lék, který přinesl v minulosti zlepšení stavu.
Plazmatická koncentrace trazodonu (při denní dávce 300 mg) byla v toxickém rozmezí, a to 1 380 ng/ml.
Již při dávce 75 mg trazodonu se tremor výrazně zmírnil, po odnětí léku vymizel zcela. Pacientka byla následující dny na své přání dimitována v kompenzovaném stavu.
Komentář:
Třes se při podávání trazodonu vyskytuje asi u 3–5 % pacientů [23]. Toxická koncentrace trazodonu může pravděpodobnost výskytu tohoto nežádoucího účinku ještě navýšit.
Kombinací trazodonu a fluoxetinu vzniká farmakodynamická interakce dvou serotonergních léčiv a jejich účinek se sčítá. Obě léčiva blokují přenašeč pro serotonin (SERT) a výsledkem interakce je nadměrná stimulace postsynaptických 5 HT2 receptorů serotoninem [13].
Dále se zde projevuje farmakokinetická interakce působením silného inhibitoru CYP2D6 a CYP3A4 fluoxetinu na metabolismus trazodonu, který je substrátem těchto dvou enzymů, což má za následek navýšení plazmatických koncentrací trazodonu. Toto bylo potvrzeno i laboratorně odběrem plazmatické koncentrace trazodonu. Naměřená plazmatická koncentrace byla 1 380 ng/ml. Terapeutické rozmezí plazmatické koncentrace trazodonu se uvádí 700–1 000 ng/ml. Alert hodnota, nad kterou by se neměla plazmatická koncentrace nacházet z důvodu rizika závažné toxicity léku, je 1 200 ng/ml [7]. Plazmatická koncentrace trazodonu u pacientky přesáhla i tuto hodnotu a nemocná jí zřejmě byla vystavena dlouhodobě.
Kazuistika 2: Interakce SSRI a warfarinu – lze podávat tyto dva léky souběžně?
Základní informace:
Žena, 31 let
Dg.: mentální anorexie, stav po závažném TS medikamenty
Epikríza:
Prvokontakt s psychiatrií, první hospitalizace. Pacientka byla do našeho zařízení přeložena z metabolické jednotky po závažném TS medikamenty (lamotrigin, venlafaxin). V rámci této hospitalizace proběhla hemodynamicky nevýznamná plicní embolie potvrzená vyšetřením výpočetní tomografií (CT), proto byla pacientka antikoagulována nízkomolekulárními hepariny (LMWH) s převodem na warfarin již před překladem (INR bylo v normě).
Po překladu do psychiatrického zařízení byla v plánu stabilizace a nastavení antidepresivní terapie, po konzultaci s psychiatrickou ambulancí bylo vybráno antidepresivum ze skupiny SSRI – fluoxetin.
Medikace při přijetí (8. 4. 2021):
pantoprazol 40 mg tbl. 1 0 0
warfarin 10 mg tbl. 1 0 0
Další vývoj:
10. 4. 2021:
Pacientka byla klidná, avšak s kolísavou úzkostí, s náhledem na TS pokus, aktivně pracovala na svých tématech na skupinové a individuální psychoterapii.
Dle plánu byla zahájena léčba antidepresivem fluoxetin a nízkou dávkou benzodiazepinů na pokrytí úzkosti, která byla zpočátku výraznější.
Somaticky: BMI 19 kg/m2
INR 2,6
Medikace:
fluoxetin 20 mg tbl. 1 0 0
klonazepam 0,5 mg tbl. 1/2 1/2 1/2
pantoprazol 40 mg tbl. 1 0 0
warfarin 10 mg tbl. 1 0 0
Po nasazení fluoxetinu začala hodnota INR postupně stoupat do supraterapeutických mezí:
Dne 17. dubna 2021 bylo podávání warfarinu na jeden den pozastaveno, další den byla dávka upravena na 5 mg/den. Bylo vyžádáno konzilium klinického farmaceuta pro posouzení lékového režimu z hlediska postupného nárůstu hodnoty INR a možnosti vlivu medikace na tento nárůst.
Konzilium klinického farmaceuta:
Na nárůstu INR se může podílet fluoxetin, který jako inhibitor CYP2D6, CYP3A4 a CYP2C19 může zvyšovat plazmatické koncentrace warfarinu, jenž je substrátem těchto izoforem. Tuto kombinaci lze však podávat v případě efektivity antidepresiva nadále za předpokladu snížení dávky warfarinu a opakované kontroly INR.
Komentář:
V tomto lékovém režimu jsou přítomny dvě farmakokinetické lékové interakce, a to na úrovni distribuce a metabolismu.
Při současném podávání warfarinu a zahájení léčby SSRI s vysokou vazbou na bílkoviny (zde fluoxetin) je potřeba počítat s tím, že může dojít k nárůstu INR, a to i mimo terapeutické rozmezí. To nastává v důsledku vytěsnění warfarinu z vazby na plazmatické bílkoviny.
Fluoxetin jako silný inhibitor CYP2D6 a středně silný inhibitor CYP3A4 a 2C19 může ovlivňovat také metabolismus warfarinu ve smyslu zvýšení jeho plazmatické koncentrace, protože warfarin je substrátem všech těchto izoforem.
Současně lze popsat
i farmakodynamickou interakci, a to zvýšení rizika
krvácení. Pokud pacient užívá současně warfarin a SSRI,
absolutní riziko krvácení do GIT je vyjádřeno 3–4 %,
i když v tomto ohledu nejsou data zcela konzistentní
[8,24].
Neznamená to však, že nelze SSRI souběžně
s warfarinem podávat. Jen je potřeba těsnější kontrola INR
a případná úprava dávky warfarinu. Obavy z uvedeného
zvýšení rizika krvácení při podávání SSRI spolu
s warfarinem by neměly vést k tomu, že lékař rezignuje
na léčbu deprese či úzkostí. Neléčená deprese či
úzkosti totiž představují pro pacienta velmi závažné riziko,
které v krajním případě může nemocného také ohrožovat
na životě [16].
Seznam použité literatury
- [1] Ereshefsky L, Stanford J, Grothe D. Antidepressant Drug‑Drug Interaction Profile Update. Drugs R D 2005; 6: 323–336.
- [2] Classen DC, Pestotnik SL, Evans RS, et al. Adverse Drug Events in Hospitalized Patients: Excess Lenght of Stay, Extra Costs, and Attributable Mortality. JAMA 1997; 277: 301–306.
- [3] Rottenkolber D, Hasford J, Stausberg J. Cost of Adverse Drug Events in German Hospitals – A Microcosting Study. Value Health 2012; 15: 868–875.
- [4] Dechanont S, Maphanta S, Butthum B, et al. Hospital admissions/visits associated with drug‑drug interactions: a systematic review and meta‑analysis. Pharmacoepidemiol Drug Saf 2014; 23: 489–497.
- [5] Gonzaga de Andrade Santos TN, Mendonça da Cruz Macieira G, Cardoso Sodré Alves BM, et al. Prevalence of clinically manifested drug interactions in hospitalized patients: A systematic review and meta‑analysis. PLoS One 2020; 15: doi:10.1371/journal.pone.0235353.
- [6] Lincová D, Farghali H. Základní a aplikovaná farmakologie. 2. vydání. Praha: Galén, 2007.
- [7] Hiemke C, Bergemann N, Clement HV, et al. AGNP Consensus Guidelines for Therapeutic Drug Monitoring in Psychiatry: Update 2017. Pharmacopsychiatry 2018; 51: 9–62.
- [8] Taylor D, Barnes TRE, Young AH. The Maudsley Prescribing Guidelines in Psychiatry. 13. vydání. London: Wiley Blackwell, 2018.
- [9] Ďuricová J, Grundmann M. CYP2D6 a jeho klinický význam. Klin Farmakol Farm 2007; 21: 133–136.
- [10] Šilhán P, Kacířová I, Hýža M, et al. Terapeutické monitorování hladin léčiv v psychiatrii – možnosti využití v praxi. Psychiatr praxi 2016; 17: 10–14.
- [11] Tašková I. Terapeutické monitorování psychofarmak z pohledu klinického farmaceuta: doporučení versus praxe. Remedia 2020; 30: 650–656.
- [12] Kršiak M. Působení léčiv na úrovni iontových kanálů a transportních bílkovin. Remedia 2014; 24: 486–490.
- [13] Stahl S. Stahl’s Essential Psychopharmacology – Neuroscientific Basis and Practical Applications. 4. vydání. Cambridge: Cambridge University Press, 2016.
- [14] Wenzel‑Seifert K, Wittmann M, Haen E. QTc Prolongation by Psychotropic Drugs and the Risk of Torsade de Pointes. Dtsch Arztebl Int 2011; 108: 687–693.
- [15] Prokeš M, Suchopár J. Lékové interakce v psychiatrii – teorie a praxe. Psychiatr praxi 2015; 16: 51–56.
- [16] Tašková I, et al. Psychofarmaka v kazuistikách. 1. vydání. Praha: Maxdorf Jessenius, 2021.
- [17] Lüllmann H, Mohr K, Wehling M. Farmakologie a toxikologie. Praha: Grada, 2014.
- [18] Vokurka M, Hugo J. Velký lékařský slovník. 4. vydání. Praha: Maxdorf, 2004.
- [19] Zanger UM, Schwab M. Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther 2013; 138: 103–141.
- [20] Tantisira K, Weiss ST. Owerview of pharmacogenomics. UpToDate. 2021. Dostupné na: www.uptodate.com.
- [21] Akamine Y, Yasui‑Furukori N, Ieiri I, et al. Psychotropic drug‑drug interactions involving P‑glycoprotein. CNS Drugs 2012; 26: 959–973.
- [22] Pechandová K, Buzková H, Slanař O, et al. Efluxní transmembránový transportér – P‑glykoprotein. Klin Biochem Metab 2006; 4: 196–201.
- [23] Trazodone, Drug Information, LexiComp. UpToDate. Dostupné na: www.uptodate.com
- [24] Cheng YL, Hu HY, Lin XH, et al. Use of SSRI, But Not SNRI, Increased Upper and Lower Gastrointestinal Bleeding: A Nationwide Population‑Based Cohort Study in Taiwan. Medicine (Baltimore) 2015; 94: e2022.