Lékové interakce cenobamátu
Souhrn:
Suchopár J, Suchopár Š, Prokeš M. Lékové interakce cenobamátu. Remedia 2022; 32: 488–495.
Cenobamát je nový protizáchvatový lék s unikátními vlastnostmi, ze kterých vyplývají jeho lékové interakce. Cenobamát se extenzivně metabolizuje především cestou UGT2B7, vyvolává středně silnou indukci CYP3A4 a slabou indukci CYP2B6 a působí jako středně silný inhibitor CYP2C19. Cenobamát zkracuje délku intervalu QT. Lékové interakce cenobamátu mohou být klinicky významné, a to jak při jeho kombinaci s jinými protizáchvatovými léky, tak i s léky používanými v běžné klinické praxi. Zajištění bezpečnosti při souběžném podávání jiných léků vede k lepší toleranci cenobamátu a k zabránění předčasnému vysazení tohoto účinného protizáchvatového léku.
Summary:
Suchopar J, Suchopar S, Prokes M. Drug interactions of cenobamate. Remedia 2022; 32: 488–495..
Cenobamate is a new antiseizure drug with unique properties that result in its drug interactions. Cenobamate is extensively metabolized primarily via the UGT2B7 pathway, induces causes moderate CYP3A4 and weak CYP2B6 induction, and acts as a moderate inhibitor of CYP2C19. Cenobamate shortens the QT interval. Drug interactions of cenobamate may be clinically significant, both when combined with other antiseizure drugs and with drugs used in routine clinical practice. Ensuring the safety of concomitant administration of other drugs leads to better tolerability of cenobamate and to the prevention of premature withdrawal of this effective antiseizure drug.
Key words: cenobamate, antiseizure medications, drug interactions, CYP3A4 induction, CYP2C19 inhibition, UGT2B7, QT shortening.
Úvodní informace
Tato práce se soustředí výhradně
na lékové interakce cenobamátu a nemá ambice hodnotit
jeho klinickou účinnost. Cenobamát je nový protizáchvatový lék
registrovaný v zemích Evropské unie dne 26. března 2021
pod názvem Ontozry
a ve Spojených státech amerických 21. listopadu
2019 pod názvem Xcopri
v indikaci přídatná léčba fokálních (parciálních)
záchvatů s generalizací nebo bez ní po selhání
předchozí terapie nejméně dvěma protizáchvatovými léky
u dospělých pacientů.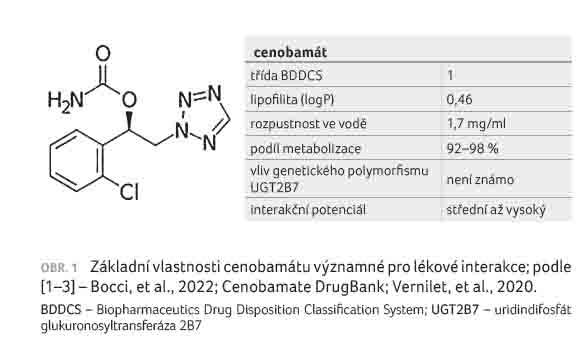
Cenobamát je malá, relativně jednoduchá molekula, chemicky se jedná o [(1R) 1 (2 chlorofenyl) 2 (tetrazol 2 yl)ethyl] karbamát, tedy levotočivý enantiomer, obrázek 1 [1–3]. Cenobamát podléhá extenzivnímu metabolismu [3], vyznačuje se nízkou lipofilitou [4] a vysokou rozpustností ve vodě [5], a proto je řazen do třídy 1 klasifikace BDDCS (The Biopharmaceutics Drug Disposition Classification System) [1]. Vliv transportních systémů na osud cenobamátu lze očekávat minimální [6], což značně zjednodušuje problematiku jeho lékových interakcí.
Cenobamát se extenzivně metabolizuje především glukuronidací, a to cestou uridindifosfát glukuronosyltransferázy 2B7 (UGT2B7) za přispění UGT2B4, glukuronidací přitom prochází jak mateřská látka, a to za vzniku N glukuronidů, tak i její oxidativní metabolity, a to za vzniku O glukuronidů [7]. N glukuronidy tvoří celkem 39,4 % metabolitů, O glukuronidy 20,4 % a nezměněný cenobamát tvoří 6,8 %. Cenobamát a jeho metabolity se eliminují převážně močí, z toho 6 % v nezměněné formě a 88 % ve formě metabolitů. Podíl eliminace stolicí je malý, vylučuje se jí pouze kolem 0,5 % mateřské látky a 5–10 % metabolitů. Všech osm známých metabolitů cenobamátu je farmakologicky neaktivních. Není známo, zda má genetický polymorfismus UGT2B7 vliv na farmakokinetické vlastnosti, účinnost nebo bezpečnost terapie cenobamátem.
Oxidativnímu metabolismu podléhá přibližně 20 % cenobamátu a děje se cestou izoenzymů cytochromu P450 CYP2E1, CYP2A6 a CYP2B6 za malého přispění CYP2C19 a CYP3A4/5. Je málo pravděpodobné, že by inhibitory, případně induktory uvedených izoenzymů cytochromu P450 vyvolávaly klinicky relevantní lékové interakce. Na druhou stranu by polyfunkční induktory cytochromu P450 a UGT, jako je rifampicin, mohly vyvolat klinicky relevantní pokles expozice cenobamátu. Není však známo, a ani nebylo nikdy modelováno, jaký důsledek by uvedené souběžné podávání cenobamátu a rifampicinu mělo.
Podání cenobamátu po jídle mírně (o zhruba 10 %) snižuje plochu pod křivkou plazmatické koncentrace cenobamátu a jeho maximální plazmatické koncentrace a vede k prodloužení času nutného k jejich dosažení. Tyto změny jsou klinicky nevýznamné, a proto je možné cenobamát podávat nezávisle na jídle, i když lze pacientům doporučit, aby cenobamát užívali pokud možno vždy ve stejnou dobu a buď nalačno, nebo po jídle.
Cenobamát má absolutní biologickou dostupnost po perorálním podání 88 %, málo se váže na plazmatické bílkoviny (kolem 60 %) a má středně velký distribuční objem 0,6–0,7 l/kg, který v zásadě odpovídá objemu tělesné vody. Biologický poločas cenobamátu je 50–60 hodin [3,8].
Cenobamát působí in vitro jako inhibitor enzymu CYP2B6 (s IC50 280 μM), CYP2C19 (s IC50 170 μM) a CYP3A4 (s IC50 720 μM). Maximální plazmatické koncentrace cenobamátu při podávání dávek 200 mg denně činí in vivo kolem 90 μM, koncentrace cenobamátu v játrech a ledvinách však mohou být významně vyšší a při dávkování 400 mg cenobamátu denně dosahují kolem 200 μM. Cenobamát lze tedy charakterizovat jako smíšený inhibitor/induktor CYP3A4 a CYP2B6, přičemž in vivo se v obou případech uplatňuje pouze jeho indukční aktivita. Inhibiční efekt cenobamátu se in vivo uplatňuje pouze v případě CYP2C19, což bylo prokázáno v cílené studii při souběžném podávání substrátu CYP2C19 omeprazolu [9], kde bylo zjištěno, že se jedná o hraničně středně silnou inhibici, neboť zvýšení plochy pod křivkou plazmatické koncentrace omeprazolu činilo 107 % (44–198 % na 90% hladině spolehlivosti [confidence interval, CI]). Cenobamát neinhibuje CYP1A2, CYP2C8, CYP2C9 ani CYP2D6 [10].
Opakované podávání cenobamátu způsobuje zvýšení exprese CYP2B6, CYP2C8 a CYP3A4. Takto navozená indukce CYP2B6 je slabá a v případě CYP2C8 je klinicky zanedbatelná. Indukce CYP3A4 je středně silná [10], což bylo prokázáno in vivo v cílené studii [9] při souběžném podávání cenobamátu s citlivým substrátem CYP3A4 midazolamem, jehož snížení plochy pod křivkou při dávkování cenobamátu 200 mg denně činilo 72 % (90% CI 68–76 %).
Z uvedených vlastností cenobamátu vyplývají jeho popsané nebo potenciální lékové interakce.
Lékové interakce s jinými protizáchvatovými léky
Cenobamát je in vivo středně silným induktorem CYP3A4 a současně je středně silným inhibitorem CYP2C19. Z těchto důvodů by cenobamát měl snižovat expozici protizáchvatovým lékům metabolizovaným cestou CYP3A4 (jako je např. karbamazepin) a snižovat jejich účinek, a naopak zvyšovat expozici protizáchvatovým lékům metabolizovaným cestou CYP2C19 (jako je např. fenobarbital, fenytoin nebo klobazam) a případně i výskyt jejich nežádoucích účinků. Cenobamát může také ovlivňovat rychlost glukuronidace protizáchvatových léků, jejichž metabolismus je závislý na glukuronidaci (jako je např. lamotrigin). Díky odlišnému mechanismu účinku také cenobamát interaguje s protizáchvatovými léky farmakodynamicky, což může vyžadovat snížení dávek. Důsledky souběžného podávání cenobamátu s jinými protizáchvatovými léky přehledně uvádí tabulka 1 [8,10,11].
Dosud provedené klinické studie
zaměřené na lékové interakce výše uvedené předpoklady
potvrdily [10]. Podávání cenobamátu zvyšuje expozici
fenobarbitalu a fenytoinu, což jsou substráty CYP2C19. Bylo
také prokázáno, že cenobamát snižuje expozici karbamazepinu,
což je substrát CYP3A4. Z protizáchvatových léků je
substrátem CYP2C19 dále brivaracetam, lakosamid, primidon,
stiripentol a kanabidiol.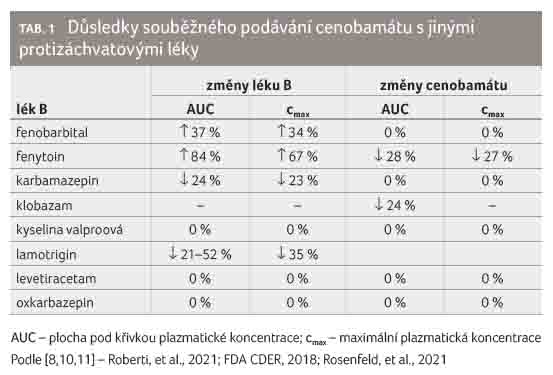
Podrobněji popíšeme důsledky souběžného podávání cenobamátu a klobazamu. Klobazam je metabolizován na aktivní metabolit N demetylklobazam (norklobazam) prostřednictvím CYP3A4, CYP2C19 a CYP2B6, N demetylklobazam je velmi citlivým substrátem CYP2C19 [12]. Souběžné podávání klobazamu s cenobamátem povede ke zvýšení tvorby N demetylklobazamu (indukcí CYP3A4) a k inhibici metabolizace tohoto aktivního metabolitu klobazamu (inhibicí CYP2C19). Dosud sice nebyla provedena cílená studie zaměřená na průkaz rozsahu změn farmakokinetických vlastností klobazamu a N demetylklobazamu, dostupné jsou ale výsledky rozsáhlé klinické studie sledující potřebu redukce dávek klobazamu a dopady změn v jeho dávkování při souběžném podávání s cenobamátem. V této studii byl při zahájení podávání cenobamátu klobazam podáván již v průměrných dávkách 38 mg denně ve skupině pacientů, kteří následně v terapii cenobamátem pokračovali, respektive v průměrných dávkách 36 mg denně ve skupině pacientů, kteří terapii cenobamátem následně ukončili [11]. V první skupině pacientů, kteří v terapii cenobamátem pokračovali, byla průměrná dávka klobazamu 38 mg denně snížena na 13 mg denně (tj. o 65,8 %), zatímco ve druhé skupině u pacientů, kteří následně terapii cenobamátem ukončili pro výskyt nežádoucích účinků, byl klobazam podáván v průměrných dávkách 36 mg denně a tato dávka byla snížena pouze na 24 mg denně (tj. o 33,3 %). Je tedy zřejmé, že (postupná) redukce dávky klobazamu souvisela s perzistencí na léčbě cenobamátem. Správná titrace dávky cenobamátu a retitrace dávky druhého protizáchvatového léku, v tomto případě klobazamu, je tak zásadní pro zajištění dostatečně dobré tolerance cenobamátu. Je velmi pravděpodobné, že při souběžném podávání cenobamátu a klobazamu dochází ke změnám farmakokinetických vlastností klobazamu, které byly pozorovány při jeho lékové interakci s kanabidiolem. Mechanismus těchto dvou lékových interakcí je totiž prakticky stejný.
Kanabidiol je, podobně jako cenobamát, středně silným inhibitorem CYP2C19. Souběžné podávání kanabidiolu s klobazamem vedlo u zdravých dobrovolníků [13] ke zvýšení plochy pod křivkou plazmatické koncentrace klobazamu o 21 % (90% CI 5−39 %) a zvýšení jeho maximálních plazmatických koncentrací o 20 % (90% CI 5−38 %), přičemž tyto změny nebyly klinicky významné. Došlo však ke zvýšení plochy pod křivkou aktivního metabolitu N desmetylklobazamu o 238 % (90% CI 162−336 %) a zvýšení jeho maximálních plazmatických koncentrací o 239 % (90% CI 161−339 %). Tyto změny lze považovat za klinicky významné, přičemž rozsah změn je důvodem pro redukci dávky klobazamu.
Také v případě souběžného
podávání fenytoinu s cenobamátem je doporučováno snížit
dávky fenytoinu, a to až o 50 %, optimálně
na podkladě monitorování jeho plazmatických koncentrací.
Doporučení o redukci dávek se týká také fenobarbitalu nebo
klobazamu. Snížení dávek uvedených protizáchvatových léků by
mělo být proaktivní (preventivní), náležitě naplánované
a nemělo by být vyčkáváno až na vznik nežádoucích
účinků nebo na výsledky terapeutického monitorování
plazmatických hladin a přijato by mělo být během titrační
fáze po zahájení podávání cenobamátu, což podrobně
popisujeme v tabulce 2 [14].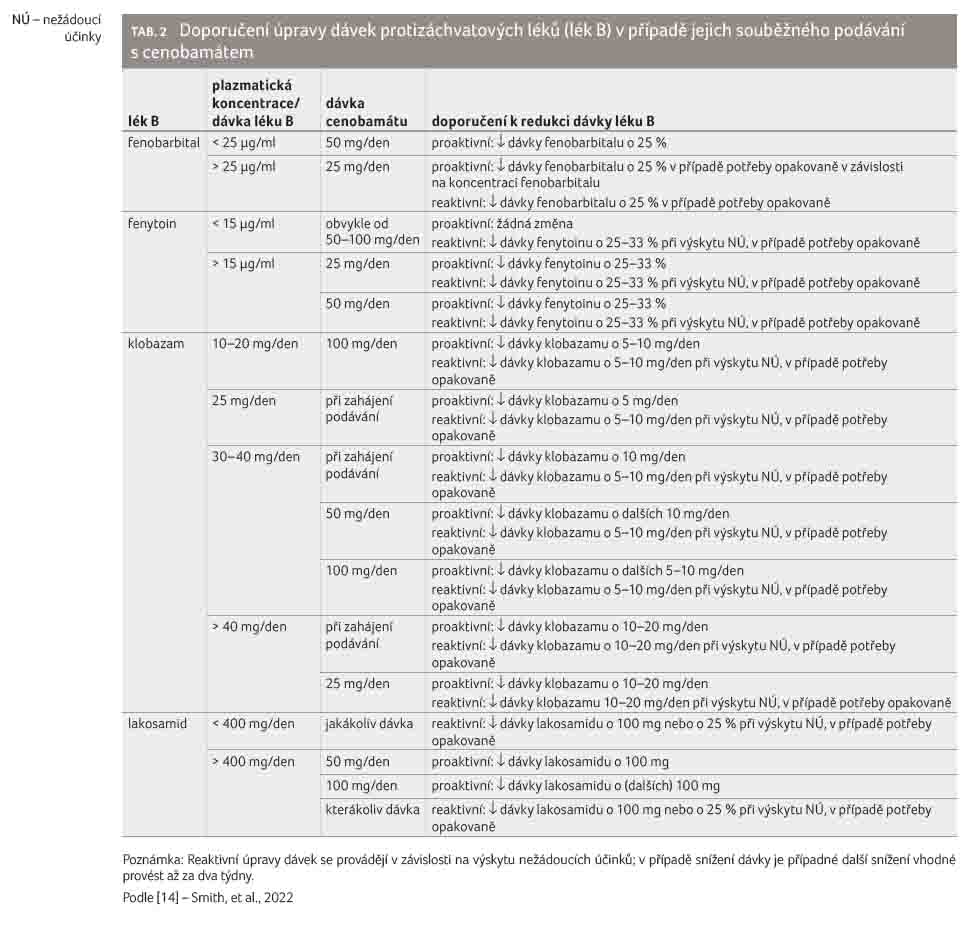
Naopak v případě souběžného podávání karbamazepinu nebo lamotriginu s cenobamátem může být potřebné zvýšit jejich dávky, i když Souhrn údajů o přípravku cenobamátu v ČR (EU) [15] uvádí, že nejsou nutné úpravy dávkování. Výjimkou je podávání vyšších dávek těchto protizáchvatových léků, tj. > 1 200 mg karbamazepinu denně nebo > 500 mg lamotriginu denně, kdy se doporučuje nepřekračovat denní dávku cenobamátu ve výši 200 mg denně. Případné změny dávek cenobamátu by měly být prováděny postupně, vždy v odstupu dvou týdnů. V případě souběžného podávání lamotriginu a cenobamátu může být také potřebné podávání vyšších dávek cenobamátu (200−400 mg denně).
V tomto smyslu vyznívají také doporučení skupiny expertů. Z těchto doporučení [14] přebíráme způsob snižování dávek v případě proaktivního (tedy preventivního) přístupu nebo při reaktivním přístupu v reakci na vznik nežádoucího účinku (tab. 2). Tabulku je třeba interpretovat následovně: např. pacient je již léčen klobazamem v dávkách 30 mg denně, ihned po zahájení podávání cenobamátu je vhodné snížit dávku klobazamu o 10 mg/den (proaktivní přístup), po dosažení dávky cenobamátu 50 mg denně je vhodné (proaktivně) snížit dávku klobazamu o dalších 10 mg denně a po dosažení dávky cenobamátu 100 mg denně je vhodné (proaktivně) snížit dávku klobazamu o dalších 5–10 mg denně.
Lékové interakce s dalšími léky
Cenobamát jako „victim“ lékových interakcí
Nejsou dostupné žádné důkazy in vitro, že by cenobamát vystupoval jako „victim“, tedy „oběť“ lékových interakcí. Cenobamát totiž není citlivým substrátem jednotlivých izoenzymů cytochromu P450. Stejně tak není citlivým substrátem jednotlivých transportních systémů. Nejsou ani publikovány žádné informace in vivo o lékových interakcích cenobamátu na podkladě inhibice nebo indukce jeho metabolismu nebo transportu.
Nelze vyloučit, že expozici cenobamátu a jeho plazmatické koncentrace zvyšují inhibitory UGT2B7, která je exprimována ve střevě, játrech a v ledvinách [16]. Ke glukuronidaci cenobamátu tak může v malé míře docházet již při procesu vstřebávání, avšak vzhledem k nízké aktivitě UGT2B7 ve střevě její případné změny dané inhibicí či indukcí patrně nebudou klinicky významné. Významně vyšší aktivita UGT2B7 je v ledvinách, kde tvoří téměř 40 % veškeré aktivity UGT [17].
Silným specifickým inhibitorem UGT2B7 je flukonazol [18], jenž je také středně silným inhibitorem CYP2C9 a silným inhibitorem CYP2C19. Souběžné podávání flukonazolu s nalbufinem, který se ze 75 % metabolizuje glukuronidací (substrát UGT2B7 za přispění UGT1A3 a UGT1A9) a z 25 % se metabolizuje oxidační hydroxylací (cestou CYP2C9 a CYP2C19), vedlo k excesivnímu zvýšení expozice nalbufinu o 1 220 % a k nárůstu jeho maximálních plazmatických koncentrací o 1 140 %, a to v důsledku silné kombinované inhibice CYP2C9, CYP2C19 a UGT2B7 [18].
Řada látek „přírodního“ charakteru působí jako silné inhibitory UGT2B7. Za zmínku jistě stojí silný inhibiční účinek steviolu [19], používaného jako sladidlo, nebo kyseliny glycyrrhizové [20], významné obsahové látky lékořice lysé (Glycyrrhiza glabra) a lékořice uralské (Glycyrrhiza uralensis), které jsou součástí naší, respektive čínské tradiční medicíny. Není známo, zda cenobamát interaguje s flukonazolem nebo s uvedenými fytofarmaky či se sladidlem steviolem, nelze to ale vyloučit.
Cenobamát jako „perpetrator“ v lékových interakcích
Cenobamát patří k lékům, které často mohou být „perpetratorem“ (vyvolavatelem) lékových interakcí. Souběžné podávání cenobamátu s midazolamem ukázalo na patrně nejvýznamnější mechanismus jeho lékových interakcí. Ve studii u zdravých dobrovolníků [9] byl po dobu 98 dnů podáván cenobamát, nejprve v dávkách 12,5 mg 1krát denně (od 1. do 14. dne), 25 mg 1krát denně (od 15. do 28. dne), 50 mg 1krát denně (od 29. do 42. dne), 100 mg 1krát denně (od 43. do 58. dne), 150 mg 1krát denně (od 59. do 72. dne) a 200 mg 1krát denně (od 73. do 98. dne). Před zahájením podávání cenobamátu, v 57. dni jeho podávání (tedy v době, kdy byla podávána dávka 100 mg 1krát denně) a v 93. dni jeho podávání (tedy v době, kdy byla podávána dávka 200 mg denně) byla perorálně podána jednorázová dávka midazolamu ve výši 2 mg (jako složka testovacího koktejlu spolu s 20 mg omeprazolu a 5 mg warfarinu). Vlivem cenobamátu v 57. dni jeho podávání došlo ke snížení plochy pod křivkou plazmatické koncentrace midazolamu o 27 % (90% CI 18−35 %) a snížení jeho maximálních plazmatických koncentrací o 27 % (90% CI 17−35 %). Vlivem cenobamátu v 93. dni jeho podávání došlo ke snížení plochy pod křivkou midazolamu o 72 % (90% CI 68−76 %) a snížení jeho maximálních plazmatických koncentrací o 61 % (90% CI 55−67 %). Snížení expozice midazolamu o 72 % je klinicky významné a může být spojeno se snížením účinku až s jeho úplnou ztrátou.
Cenobamát jako středně silný induktor CYP3A4 [9] ovlivňuje nebo by mohl ovlivňovat osud značného množství substrátů. To se týká např.:
- kombinovaných perorálních kontraceptiv, perorálních progestinových kontraceptiv (tzv. minipills),
- některých statinů (simvastatin, lovastatin, atorvastatin),
- dihydropyridinových kalciových blokátorů (felodipin, amlodipin, lerkanidipin),
- glukokortikoidů (systémové, jako je metylprednisolon, inhalační nebo nazální, jako je flutikason, nebo lokální, jako je beklometason),
- některých benzodiazepinů (midazolam, alprazolam),
- imunosupresiv (cyklosporin, takrolimus, everolimus),
- „klasických“ protinádorových léků (např. irinotekan, vinkristin),
- nových protinádorových léků ze skupiny inhibitorů tyrozinkinázy (např. imatinib, bosutinib, ceritinib, lorlatinib nebo tofacitinib),
- a řady dalších.
V některých případech lze lékovou interakci „vyřešit“ zvýšením dávky substrátu, např. atorvastatinu, v jiných případech, např. v případě kombinovaných perorálních kontraceptiv, to není možné. Optimálním řešením je volit takové alternativy léků, které nejsou substráty CYP3A4, např. namísto alprazolamu zvolit oxazepam, namísto atorvastatinu zvolit rosuvastatin atd.
Cenobamát je také středně silným inhibitorem CYP2C19 [9], což je druhý nejvýznamnější mechanismus lékových interakcí cenobamátu. Při podávání cenobamátu zdravým dobrovolníkům se stejnou titrací popsanou výše byla před zahájením podávání cenobamátu, v 57. a 93. dni jeho podávání perorálně podána jednorázová dávka omeprazolu ve výši 40 mg. Vlivem cenobamátu v 93. dni jeho podávání (v dávkách 200 mg 1krát denně) došlo ke zvýšení plochy pod křivkou omeprazolu o 107 % (90% CI 44–198 %) a zvýšení jeho maximálních plazmatických koncentrací o 83 % (90% CI 26–167 %). Tato léková interakce patrně není klinicky významná, neboť omeprazol je lék s velkou terapeutickou šířkou a dvojnásobné zvýšení expozice tomuto léku nepředstavuje zásadní riziko. Jiná je situace u substrátů CYP2C19 s úzkou terapeutickou šířkou, jako je např. citalopram, escitalopram, fenytoin, gliklazid, moklobemid, klopidogrel nebo vorikonazol. U takových léků by případné dvojnásobné zvýšení expozice mohlo vést ke vzniku nežádoucích účinků, nebo i toxicity.
Speciální situace by pak mohla nastat v případě souběžného podávání cenobamátu a klopidogrelu. Držitel rozhodnutí o registraci cenobamátu doporučuje: „Při souběžném podávání s cenobamátem může být nutné snížit dávky léků metabolizovaných pomocí CYP2C19“ [15]. To však pro klopidogrel neplatí. Klopidogrel se totiž cestou CYP2C19 metabolizuje na aktivní metabolit, který je vlastním nositelem protidestičkového účinku. Souběžné podávání klopidogrelu s inhibitory CYP2C19, jako je cenobamát, by mohlo vést ke snížení protidestičkového účinku.
Zkrácení intervalu QT způsobené cenobamátem je mírné a závislé na dávce cenobamátu. V souvislosti s podáváním cenobamátu však dosud nebylo pozorováno zkrácení intervalu QT pod 340 ms [15]. Možnost lékové interakce založené na mechanismu zkrácení intervalu QT je tak spíše v rovině teoretické.
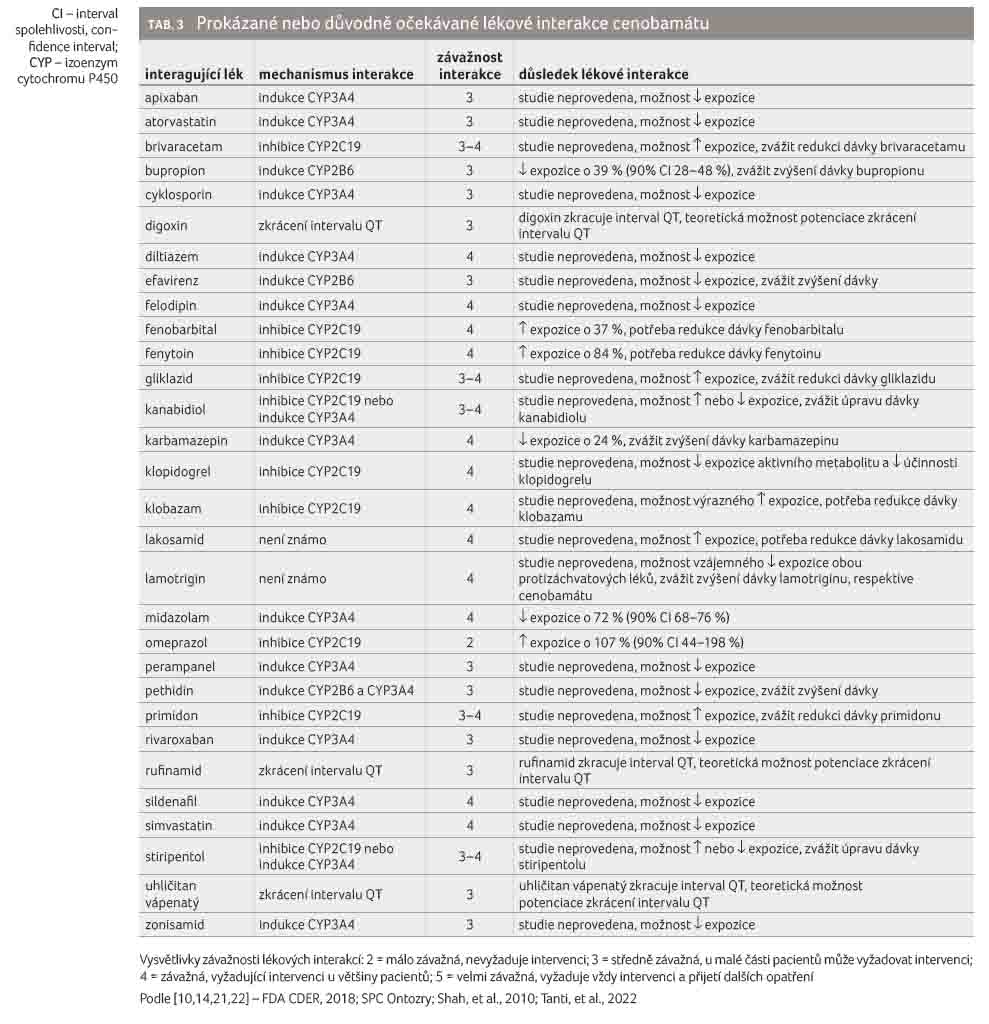
Přehled prokázaných nebo důvodně očekávaných lékových interakcí cenobamátu uvádí tabulka 3 [10,14,21,22].
{kind=link}
Seznam použité literatury
- [1] Bocci G, Oprea TI, Benet LZ. State of the Art and Uses for the Biopharmaceutics Drug Disposition Classification System (BDDCS): New Additions, Revisions, and Citation References. AAPS J 2022; 24: 37.
- [2] Cenobamate DrugBank. Dostupné na: https://go.drugbank.com/drugs/DB06119
- [3] Vernillet L, Greene SA, Kamin M. Pharmacokinetics of Cenobamate: Results From Single and Multiple Oral Ascending‑Dose Studies in Healthy Subjects. Clin Pharmacol Drug Dev 2020; 9: 428–443.
- [4] Odi R, Bibi D, Wager T, Bialer M. A perspective on the physicochemical and biopharmaceutic properties of marketed antiseizure drugs–From phenobarbital to cenobamate and beyond. Epilepsia 2020; 61: 1543–1552.
- [5] FDA Full Prescribing Information Xcopri® (cenobamate), SK Life, 6/2022. Dostupné na: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/212839s006s007lbl.pdf
- [6] Wu CY, Benet LZ. Predicting Drug Disposition via Application of BCS: Transport/Absorption/ Elimination Interplay and Development of a Biopharmaceutics Drug Disposition Classification System. Pharmaceutical Research 2005; 22: 11–23.
- [7] Vernillet L, Greene SA, Kim HW, et al. Mass Balance, Metabolism, and Excretion of Cenobamate, a New Antiepileptic Drug, After a Single Oral Administration in Healthy Male Subjects. Eur J Drug Metab Pharmacokinet 2020; 45: 513–522.
- [8] Roberti R, de Caro C, Iannone LF, et al. Pharmacology of Cenobamate: Mechanism of Action, Pharmacokinetics, Drug‑Drug Interactions and Tolerability. CNS Drugs 2021; 35: 609–618.
- [9] Greene SA, Kwak C, Kamin M, et al. Effect of cenobamate on the single‑dose pharmacokinetics of multiple cytochrome P450 probes using a cocktail approach in healthy subjects. Clin Transl Sci 2022; 15: 899–911.
- [10] FDA CDER, Clinical Pharmacology and Biopharmaceutics Reviews, Application Number 212839Orig1s000, Cenobamate, 11/2018, SK Life Science. Dostupné na: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/212839Orig1s000ClinPharmR.pdf
- [11] Rosenfeld WE, Abou‑Khalil B, Aboumatar S, et al. Post hoc analysis of a phase 3, multicenter, open‑label study of cenobamate for treatment of uncontrolled focal seizures: Effects of dose adjustments of concomitant antiseizure medications. Epilepsia 2021; 62: 3016–3028.
- [12] Huddart R, Leeder JS, Altman RB, et al. PharmGKB summary: clobazam pathway, pharmacokinetics. Pharmacogenet Genomics 2018; 28: 110–115.
- [13] Geffrey AL, Pollack SF, Bruno PL, Thiele EA. Drug‑drug interaction between clobazam and cannabidiol in children with refractory epilepsy. Epilepsia 2015; 56: 1246–1251.
- [14] Smith MC, Klein P, Krauss GL, et al. Dose Adjustment of Concomitant Antiseizure Medications During Cenobamate Treatment: Expert Opinion Consensus Recommendations. Neurol Ther 2022; Sep 3, doi: 10.1007/s40120‑022‑00400‑5 [online ahead of print].
- [15] SPC Ontozry. Dostupné na: https://www.ema.europa.eu/en/documents/product‑information/ontozry‑epar‑product‑information_cs.pdf
- [16] Wang H, Cao G, Wang G, Hao H. Regulation of Mammalian UDP‑Glucuronosyltransferases. Curr Drug Metab 2018; 19: 490–501.
- [17] Kasteel EEJ, Darney K, Kramer NI, et al. Human variability in isoform‑specific UDP‑glucuronosyltransferases: markers of acute and chronic exposure, polymorphisms and uncertainty factors. Arch Toxicol 2020; 94: 2637–2661.
- [18] Liang R‑J, Shih Y‑N, Chen Y‑L, et al. A dual system platform for drug metabolism: Nalbuphine as a model compound. Eur J Pharm Sci 2020; 141: 105093.
- [19] Wang M, Lu J, Li J, et al. Steviol glucuronidation and its potential interaction with UDP‑glucuronosyltransferase 2B7 substrates. Food Chem Toxicol 2014; 64: 135–143.
- [20] Huang Y‑P, Cao Y‑F, Fang Z‑Z, et al. Glycyrrhetinic acid exhibits strong inhibitory effects towards UDP‑glucuronosyltransferase (UGT) 1A3 and 2B7. Phytother Res 2013; 27: 1358–1361.
- [21] Shah RR. Drug‑induced QT interval shortening: potential harbinger of proarrhythmia and regulatory perspectives. Br J Pharmacol 2010; 159: 58–69.
- [22] Tanti A, Micallef B, Szijj JV, et al. QT shortening: a proarrhythmic safety surrogate measure or an inappropriate indicator of it? Curr Med Res Opin 2022; 38: 1473–1483.