Lékové interakce léčiv používaných v revmatologii
Souhrn:
Suchopár J, Prokeš M, Suchopár Š. Lékové interakce léčiv používaných v revmatologii. Remedia 2019; 29: 481–491.
Lékové interakce se v současné době poměrně běžně vyskytují jak při poskytování ambulantní péče, tak i u hospitalizovaných pacientů. Výskyt lékových interakcí je v podmínkách České republiky častější u žen a roste s věkem pacientů, což souvisí s počtem užívaných léčiv. V oboru revmatologie se standardně používají léčiva, která mají vysoký interakční potenciál, jako je např. metotrexát nebo leflunomid. Přehledný článek podává informaci o hlavních mechanismech lékových interakcí chorobu modifikujících antirevmatických léků (disease‑modifying antirheumatic drugs, DMARDs), včetně biologických léčiv.
Summary:
Suchopar J, Prokes M, Suchopar S. Interactions of drugs in rheumatology. Remedia 2019; 29: 481–491.
Drug interactions are currently relatively common both in outpatient care and in hospitalized patients. The occurrence of drug interactions in the Czech Republic is more frequent among women and increases with the age of patients which is related to the number of drugs used. In the field of rheumatology, drugs that have a high interaction potential, such as methotrexate or leflunomide, are commonly used. The review article provides information on the main mechanisms of drug interactions of disease‑modifying antirheumatic drugs (DMARDs), including biological drugs.
Key words: drug interactions , DMARDs , biological drugs , rheumatology ,methotrexate , sulfasalazine , hydroxychloroquine , penicillamine , leflunomide ,azathioprine , ciclosporin , apremilast , tofacitinib , baricitinib , etanercept adalimumab , infliximab , certolizumab , golimumab , abatacept ,canakinumab ,secukinumab , rituximab , belimumab , anakinra , tocilizumab , sarilumab ,sirucumab.
Úvod
Popisovat lékové interakce léčiv používaných v revmatologii je složité, neboť tato skupina léčiv zahrnuje řadu různorodých podskupin, v nichž se uplatňují prakticky všechny známé mechanismy lékových interakcí. Lékové interakce léčiv používaných v revmatologii mají pochopitelně značně rozdílnou klinickou závažnost, přičemž její konkrétní míra u konkrétního pacienta je mnohdy spojena s genetickým polymorfismem, který až na výjimky není standardní součástí vyšetření nemocných. Jako v jiných oblastech i zde platí, že lékové interakce relativně málo závažné mají vysokou incidenci a mohou být populačně nebezpečnější než závažné lékové interakce s minimálním výskytem.
Léčiva používaná v revmatologii jsou pro účely tohoto přehledu rozdělena na:
a) nesteroidní protizánětlivá léčiva (NSA)
- cyklooxygenáza 2 (COX 2) neselektivní se spotřebou 57,19 definované denní dávky na 1 000 obyvatel a den (DDD/TID) ‒ např. diklofenak, ibuprofen, ale i meloxikam
- COX 2 selektivní se spotřebou 0,30 DDD/TID – např. celekoxib
b) syntetické konvenční chorobu modifikující antirevmatické léky (conventional synthetic disease modifying antirheumatic drugs, csDMARDs)
- metotrexát se spotřebou 2,65 DDD/TID
- sulfasalazin se spotřebou 0,77 DDD/TID
- hydroxychlorochin se spotřebou 0,44 DDD/TID
- penicilamin se spotřebou 0,05 DDD/TID
- leflunomid se spotřebou 0,47 DDD/TID
- azatioprin se spotřebou 0,90 DDD/TID
- cyklosporin se spotřebou 0,20 DDD/TID
c) cílené syntetické DMARDs (targeted synthetic DMARDs, tsDMARDs)
- apremilast se spotřebou 0,002 DDD/TID
- tofacitinib se spotřebou 0,001 DDD/TID
- baricitinib se spotřebou 0,001 DDD/TID
d) biologické DMARDs (biologic DMARDs, bDMARDs)
- inhibitory tumor nekrotizujícího faktoru alfa (TNFα) se spotřebou 1,40 DDD/TID ‒ etanercept, adalimumab, infliximab, certolizumab a golimumab
- inhibitory interleukinu 1 (IL 1) se spotřebou 0,0042 DDD/TID ‒ anakinra, kanakinumab
- inhibitory interleukinu 6 (IL 6) se spotřebou 0,0634 DDD/TID ‒ tocilizumab, sarilumab, sirukumab
- inhibitory interleukinu 17A (IL 17A) se spotřebou 0,0213 DDD/TID ‒ sekukinumab
- léčiva s jiným mechanismem se spotřebou 0,0154 DDD/TID ‒ abatacept, rituximab, belimumab
Všechna NSA mají schválenou indikaci symptomatická léčba bolesti při akutní artritidě a řadu dalších indikací u bolestivých projevů zánětlivých onemocnění. Pouze lornoxikam, dexketoprofen, nimesulid a parekoxib lze podle Souhrnu údajů o přípravku (SPC) využít jen jako analgetikum.
Revmatoidní artritidu mají v platném SPC schválenou všechna csDMARDs, ze skupiny tsDMARDs pak tofacitinib a baricitinib (apremilast má schválenou indikaci psoriatická artritida a psoriáza) a ze skupiny bDMARDs mají indikaci revmatoidní artritida schválenou všechny inhibitory TNFα, anakinra, tocilizumab, sarilumab, sirukumab, abatacept a rituximab. Kanakinumab má schválenou indikaci Stillova choroba a dnavá artritida, sekukinumab má schválenou indikaci ankylozující spondylitida, psoriatická artritida a psoriáza a belimumab má schválenou indikaci systémový lupus erythematodes.
V případě léčiv používaných v revmatologii v mechanismech lékových interakcí převažují farmakodynamické lékové interakce. Jedná se zejména o zesílení hepatotoxicity, nefrotoxicity či útlum krvetvorby, což jsou vlastnosti, které jsou typické pro většinu csDMARDs. Je však nezbytné uvést, že tyto lékové interakce jsou obvykle málo závažné a jejich výskyt (možná překvapivě) není příliš vysoký. Hydroxychlorochin, jako ostatní deriváty 4 aminochinolinu, prodlužuje korigovaný interval QT (QTc) a může tak vyvolat komorové arytmie typu torsade de pointes.
Farmakokinetické lékové interakce tvoří sice menší, ale o to důležitější část pozorovaných lékových interakcí. Jak bude podrobně uvedeno v dalším textu, bývají zejména csDMARDs a tsDMARDs substráty různých biotransformačních enzymů I. nebo II. fáze biotransformace (jako je např. cytochrom P450 3A4 ‒ CYP3A4 ‒ nebo glukuronyltransferáza) a jsou též substráty řady transportních systémů (jako je např. P glykoprotein).
V případě řady bDMARDs může docházet k lékovým interakcím velmi specifickým způsobem, který je dán vyššími koncentracemi prozánětlivých cytokinů u dosud neléčených nebo nedostatečně léčených pacientů. Zejména vyšší koncentrace IL 1 nebo IL 6 vedou ke snížení exprese biotransformačních enzymů I. fáze biotransformace. U pacientů v důsledku chronického zánětlivého onemocnění dochází ke změně fenotypu na pomalého nebo středně rychlého metabolizátora, přičemž po zahájení aplikace biologické terapie na bázi inhibice IL 1 a především IL 6 se navodí stav, kdy se normalizuje exprese např. CYP3A4 na původní úroveň, což ale znamená klinicky významné zvýšení aktivity CYP3A4. Dosud užívané dávky substrátů CYP3A4 již nemusejí stačit k vyvolání totožné odpovědi před biologickou léčbou.
Přehled vlastností důležitých pro
lékové interakce u léčiv používaných v revmatologii
Všechna léčiva ze skupiny csDMARDs jsou substráty některého z transportních systémů a některá z nich jsou pak substrátem biotransformačních enzymů I. fáze biotransformace, zejména CYP450 (cyklosporin, leflunomid a hydroxychlorochin) nebo II. fáze biotransformace (sulfasalazin, cyklosporin).
Některá csDMARDs působí jako
inhibitor různých enzymů (např. leflunomid je středně silným
inhibitorem CYP2C8) nebo jako induktor (např. leflunomid je slabým
induktorem CYP1A2). Konvenční syntetické DMARDs působí též
jako inhibitor různých transportních systémů (např. leflunomid
je polyfunkčním středně silným inhibitorem OATP1B1, OATP1B3,
OAT3, MRP2, MRP4 a BCRP), tabulka 1 [1].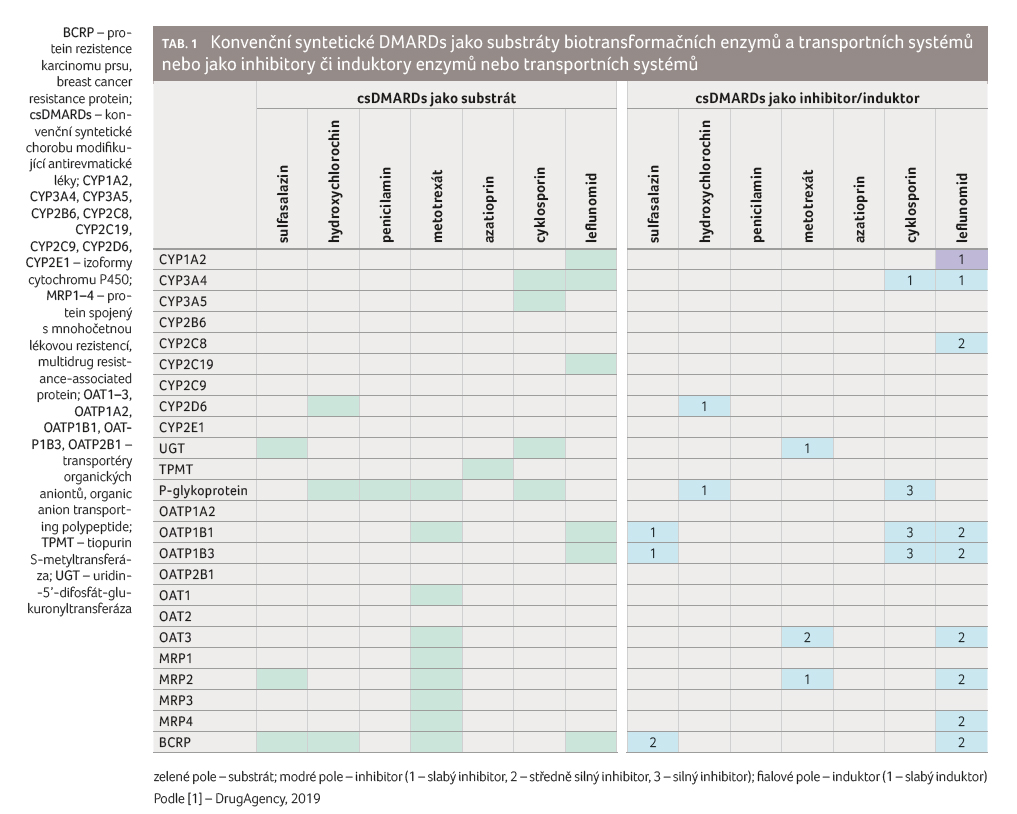
Interpretace údajů v tabulce je jednoduchá. Jestliže je csDMARD substrátem enzymu, např. hydroxychlorochin CYP2D6, pak silné inhibitory CYP2D6 (např. paroxetin, fluoxetin nebo bupropion) zvyšují jeho expozici a případně i výskyt nežádoucích účinků. Je li csDMARD substrátem transportního systému, pak inhibitory tohoto transportního systému buď zvyšují expozici, a to zamezením funkce efluxních transportních systémů, jako je P glykoprotein nebo BCRP v gastrointestinálním traktu, nebo zvyšují expozici zamezením funkce influxních transportních systémů (např. OATP1B1, AOT3, MRP2, MRP4) v ledvinách nebo v játrech. Induktory transportních systémů působí opačně, tj. vedou ke snížení expozice a tím i účinku csDMARDs. Například podávání extraktu z třezalky tečkované povede ke snížení expozice hydroxychlorochinu, penicilaminu, metotrexátu nebo cyklosporinu (důkaz je v současné době dostupný pouze pro cyklosporin [2]).
Často jsou diskutovány otázky
vzájemného kombinování csDMARDs. Této problematice je věnována
pozornost též v aktuálních doporučených postupech
diagnostiky a léčby revmatoidní artritidy [3]. Zde uvedené
informace lze z hlediska lékových interakcí pouze potvrdit
(tab. 2).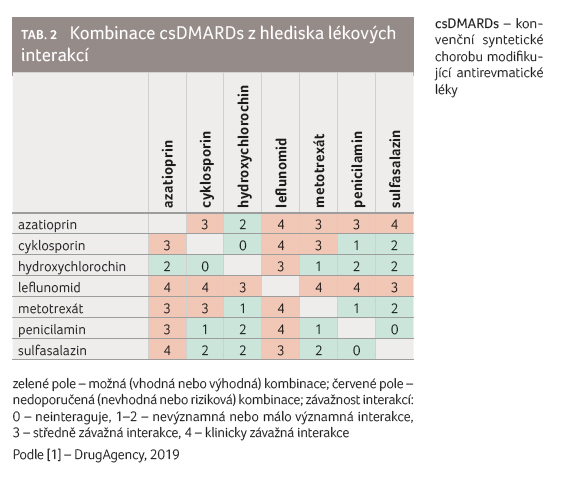
Podávat lze dvojkombinace (např. metotrexát a hydroxychlorochin) nebo trojkombinace (metotrexát, sulfasalazin a hydoxychlorochin), takové kombinace mohou být výhodné, avšak diskuse nad jejich účinností není předmětem tohoto pojednání a autoři odkazují na metaanalýzu studií [4], respektive na národní doporučené postupy [3].
Z hlediska provedených studií lékových interakcí je nezbytné zejména zmínit, že sulfasalazin je in vitro i in vivo inhibitorem RFC (přenašeč pro redukovaný folát, reduced folate carrier) a PCFT (na proton vázaný folátový transportér, proton coupled folate transporter), tedy transportních systémů pro folát, které transportují též metotrexát [5,6]. Sulfasalazin tak může ovlivnit vstřebávání perorálně podaného metotrexátu a současně může změnit jeho průnik do buněk i při parenterální aplikaci. Dále je vhodné upozornit, že nejsou k dispozici dostatečné údaje o bezpečnosti kombinací csDMARDs vyvolávajících klinicky významnou leukopenii, a proto nejsou takové kombinace doporučovány (např. cyklosporin a azatioprin, leflunomid a cyklosporin nebo azatioprin a metotrexát). V praxi se však lze často setkat s kombinací metotrexát a leflunomid, jejíž podávání výrobce leflunomidu nedoporučuje. Tato kombinace byla zkoumána z hlediska bezpečnosti ‒ např. ve studii u 30 pacientů s revmatoidní artritidou, kteří byli dlouhodobě léčeni metotrexátem v průměrných dávkách 17,2 mg týdně, byl po dobu 52 týdnů podáván též leflunomid v dávkách 10‒20 mg denně [7]. U 7 % pacientů došlo k elevaci hodnot aspartátaminotransferázy (AST) a u 17 % pacientů k elevaci hodnot alaninaminotransferázy (ALT) nad trojnásobek normálních hodnot. U 13 pacientů bylo třeba redukovat dávky leflunomidu, u tří z nich již nebylo možno terapii leflunomidem znovu zahájit. Leflunomid však nezměnil maximální plazmatické koncentrace ani plochu pod křivkou plazmatických koncentrací metotrexátu. To však neznamená, že souběžné podávání leflunomidu nemá vliv na farmakokinetické vlastnosti metotrexátu, neboť podávání leflunomidu mění distribuci metotrexátu a zvyšuje jeho koncentrace v játrech [8].
Všechna tsDMARDs jsou substráty CYP3A4. Z hlediska souběžného podávání s inhibitory CYP3A4 je však třeba intervence pouze v případě tofacitinibu, neboť celková expozice se současným podáváním silných inhibitorů CYP3A4 u apremilastu ani baricitinibu prakticky nemění. U tofacitinibu silné inhibitory CYP3A4 zvyšují expozici přibližně o 100 %, a proto výrobce doporučuje v případě souběžného podávání silných inhibitorů CYP3A4 snížit dávky tofacitinibu z obvyklých 5 mg dvakrát denně na 5 mg jednou denně. Odlišná je situace v případě kombinace tsDMARDs s induktory CYP3A4. Silné induktory snižují expozici apremilastu o 72 %, baricitinibu o 34 % a tofacitinibu o 84 %. Výrobci apremilastu a tofacitinibu proto doporučují se současnému podávání silných induktorů CYP3A4 vyhnout. Snížení expozice baricitinibu působením silného induktoru CYP3A4 je malé (jak bylo uvedeno výše, činí 34 %) a není doprovázeno změnou plazmatických koncentrací baricitinibu, proto výrobce nedoporučuje přijetí jakýchkoliv opatření z hlediska opatrnosti nebo změn v dávkování.
Při terapii tsDMARDs je třeba pacienta poučit, aby se vyhýbal přírodním léčivům, která mají klinicky významný indukční efekt. Příkladem takových léčiv je třezalka tečkovaná, šalvěj červekořenná, čajovec kapský nebo ostružiník sladký.
Jak již bylo uvedeno výše, platí pro skupinu bDMARDs, že inhibitory IL 1 a IL 6 (a nelze vyloučit, že i inhibitory TNFα) obnovují chronickým zánětem navozené snížení exprese biotransformačních enzymů I. fáze. Tato léčiva (anakinra, kanakinumab, tocilizumab, sarilumab a sirukumab) tak krátce po zahájení terapie mohou zvýšit aktivitu jednotlivých izoenzymů CYP450. To má za následek snížení expozice současně podávaným léčivům, která jsou substráty těchto enzymů. Týká se to zejména CYP3A, přičemž pro tento izoenzym jsou k dispozici dostatečné důkazy o zvýšení enzymové aktivity terapií onemocnění.
Při zahajování terapie bDMARDs je třeba opatrnosti u pacientů, kteří užívají citlivé substráty CYP3A4, CYP2C9, CYP2C19 nebo CYP1A2 s úzkým terapeutickým oknem. Je nutné sledovat účinnost těchto léčiv a výskyt nežádoucích účinků a v případě potřeby upravit dávky těchto léčiv. Takzvané rizikové období pro změnu exprese je krátké, obvykle do dvou měsíců, poté již žádné změny farmakokinetických vlastností souběžně užívaných léčiv nejsou očekávány.
Lékové interakce jednotlivých
csDMARDs
Metotrexát
Lékem první volby při terapii revmatoidní artritidy je metotrexát, který je klasickým csDMARD. Skutečnost, že se jedná o lék první volby, potvrzuje jeho rostoucí spotřeba, která v roce 2018 činila 2,88 DDD/1 000 obyvatel/den, což reprezentovalo 23% nárůst ve srovnání s rokem 2016. Původně byl metotrexát vyvinut jako antimetabolit folátů a v této indikaci byl od roku 1947 používán při terapii nádorových onemocnění. Při terapii revmatoidní artritidy se začal uplatňovat od 50. let minulého století. Podává se nejčastěji podkožně, méně často perorálně, v obou případech obvykle jednou týdně. Metotrexát patří k léčivům, která jsou poměrně často nesprávně užívána, nejčastěji pro nepochopení režimu dávkování ze strany pacienta [9].
Metotrexát patří mezi několik málo léčiv, u nichž je v databázi amerického Úřadu pro kontrolu potravin a léčiv (Food and Drug Administration, FDA) uvedeno více než 100 000 hlášení nežádoucích účinků [10]. Z toho 2 539 hlášení popisuje lékové interakce metotrexátu, z nichž 319 mělo fatální zakončení. Při terapii metotrexátem lze tedy doporučit obezřetnost s ohledem na výběr další medikace pacienta.
Po perorálním podání se metotrexát vstřebává aktivním transportem za účasti transportního systému RFC a PCFT. Tyto transportní systémy mohou být inhibovány některými léčivy, např. sulfasalazinem [5,6,11] či dolutegravirem [12]. Podání metotrexátu po jídle vedlo v některých studiích ke snížení jeho biologické dostupnosti [13,14], v jiných studiích [15,16] vliv potravy prokázán nebyl. Výrobci se shodují, že by měl být metotrexát podáván nalačno, tj. jednu hodinu před jídlem nebo dvě hodiny po něm.
Metotrexát se v organismu
částečně metabolizuje na 7 hydroxymetotrexát a z organismu
se eliminuje převážně močí, a to aktivním transportem
za účasti řady transportních systémů (tab. 4). Jedná se především o OAT3 [17], který
zajišťuje vstup metotrexátu do tubulární buňky,
a MRP2/MRP4 [18,19], které zajišťují vypuzení metotrexátu
z této buňky do primární moči. Pro zájemce o pochopení
mechanismů transportů léčiv a jejich eliminaci ledvinami lze
doporučit excelentně zpracovaný přehled [20].
Ovlivnění transportu metotrexátu při jeho eliminaci ledvinami je podstatou většiny jeho farmakokinetických lékových interakcí. Inhibitory MRP2/MRP4 jsou např. glukuronidy NSA, mj. diklofenaku nebo ibuprofenu [18]. Tytéž metabolity NSA inhibují OAT1 a OAT3 [17]. Známým inhibitorem OAT3, MRP2 a BCRP je famotidin, inhibitory protonové pumpy jsou též inhibitory OAT3, MRP4 a současně kompetují s metotrexátem při transportu cestou BCRP [21]. Zatímco v případě inhibitorů protonové pumpy dochází k lékové interakci s metotrexátem (zpoždění jeho eliminace) pravděpodobně pouze při aplikaci jeho vysokých dávek používaných v onkologii, je souběžné podávání metotrexátu s NSA zatíženo rizikem i při nízkých dávkách používaných v revmatologii [22]. Dokladem mohou být výsledky dánské studie, kde byl za období let 2004‒2015 sledován výskyt nežádoucích příhod (zejména nefrotoxicita, hepatotoxicita, myelotoxicita) u pacientů léčených metotrexátem v monoterapii (n = 21 725) nebo metotrexátem v kombinaci s NSA (n = 21 536) [23]. Současné podávání metotrexátu a NSA bylo spojeno se zvýšením relativního rizika vzniku závažné nežádoucí příhody na 1,40 (1,07‒1,82 na 95% hladině spolehlivosti). Sekundární analýzy prokázaly, že současné užívání metotrexátu a NSA bylo statisticky významně spojeno s akutním selháním funkce ledvin a s cytopenií.
Kombinace metotrexátu s kyselinou acetylsalicylovou podávanou v nízkých dávkách jako antiagregans je podle vyjádření výrobců kontraindikována, je li aplikovaná dávka metotrexátu vyšší než 15 mg týdně (Acetylsalicylic Acid Krka, Kardegic, Manoass, Stacyl, Vasopyrin), respektive pokud činí 15 mg týdně nebo více (Anopyrin, Aspirin Protect, Carsaxa, Godasal, Kyselina acetylsalicylová Xantis). Je opravdu pozoruhodné, že v případě některých obdobných přípravků s kyselinou acetylsalicylovou kombinace kontraindikovaná není (Cardioral). Ještě pozoruhodnější je, že přípravky, které dokonce obsahují vyšší (analgetické) dávky, takové omezení nemají (např. Acygal nebo Acylpyrin), přičemž ostatní léčivé přípravky s kyselinou acetylsalicylovou jsou s metotrexátem podávaným v dávkách vyšších než 15 mg týdně kontraindikovány (včetně přípravku Acylpyrin šumivé tablety). Jak se v takových informacích má lékař vyznat, je skutečně záhadou.
Potenciál lékové interakce na úrovni inhibice transportního systému OAT3 je znám ze studie lékové interakce metotrexát‒probenecid. Ve studii u pacientů s nádorovými onemocněními perorální i injekčně aplikovaný probenecid zvýšil plochu pod křivkou metotrexátu o 127 % a jeho maximální plazmatické koncentrace o 344 % [24]. Totéž bylo prokázáno u zdravých dobrovolníků, přičemž probenecid byl podáván opakovaně a plocha pod křivkou metotrexátu se zvýšila o 100 %, stejně tak i plazmatické koncentrace metotrexátu [25].
Potenciál lékové interakce na úrovni inhibice transportního systému MRP2/MRP4 je dosud znám pouze z experimentálních studií. Například reveň dlanitá (známá spíše jako rebarbora), respektive její extrakt obsahující antraglykosidy, byl potkanům podáván v dávkách 0,5 g/kg, respektive 1 g/kg spolu s metotrexátem [26]. Došlo ke zvýšení plochy pod křivkou metotrexátu o 307 %, respektive o 602 %. Jaký případný význam má tato interakce v humánní medicíně, nelze v současnosti blíže upřesnit. Studie s rebarborou však není ojedinělou informací. Další experimentální studie u potkanů prokázala zvýšení plochy pod křivkou metotrexátu při jeho současném podání se šišákem bajkalským (Scutellaria baicalensis, což je jedna z významných součástí tradiční čínské medicíny) o 242‒501 %, v závislosti na podané dávce extraktu ze šišáku [27].
Metotrexát je substrátem transportního systému BCRP, přičemž tento transportní systém ovlivňuje jak biologickou dostupnost perorálně podaného metotrexátu (exprese BCRP v enterocytu), tak i jeho eliminaci (exprese BCRP v ledvinách) [28]. Metotrexát naštěstí nemá vliv na expresi BCRP [29]. U pacientů se zvýšenou aktivitou BCRP (tj. s genetickým polymorfismem) však může dojít ke snížení účinku metotrexátu [30]. V případě perorálního podání metotrexátu může jeho biologickou dostupnost zvýšit souběžné podání inhibitoru BCRP, např. kurkuminu, podobně jako u sulfasalazinu [31]. V případě podkožní aplikace metotrexátu mohou jeho eliminaci zpozdit systémově působící inhibitory BCRP, jako je např. dasatinib [32]. Nově bylo zjištěno, že metotrexát je substrátem OATP1B1 [33].
Aplikace metotrexátu nemá vliv na farmakokinetické vlastnosti bDMARDs, stejně tak bDMARDs neovlivňují farmakokinetické vlastnosti metotrexátu. Naopak souběžné podávání metotrexátu a bDMARDs je výhodné, neboť účinek kombinace je vyšší a riziko vzniku neutralizačních protilátek nižší.
Sulfasalazin
Sulfasalazin je další csDMARD
používaný při terapii revmatoidní artritidy od 50. let
minulého století. Jedná se o dobře účinné léčivo, jehož
podávání však může být spojeno s řadou nežádoucích
účinků a také lékových interakcí (tab. 5). Významné jsou přitom právě vztahy mezi lékovými
interakcemi a nežádoucími účinky, protože v případě
sulfasalazinu se obě tyto entity prolínají. Sulfasalazin je
poměrně citlivým substrátem BCRP, proto se dokonce užívá jako
momentálně nejvhodnější modelové léčivo pro průkaz lékových
interakcí na úrovni intestinální inhibice nebo indukce BCRP
v rámci studií lékových interakcí, ačkoliv to doporučení
Evropské lékové agentury (European Medicines Agency, EMA) z roku
2012 ani FDA z roku 2017 dosud výslovně neuvádějí [34,35].
Známo je velké množství inhibitorů BCRP, přičemž se většinou
jedná o novější léčiva, ale také o dlouho známé
přírodní látky [36].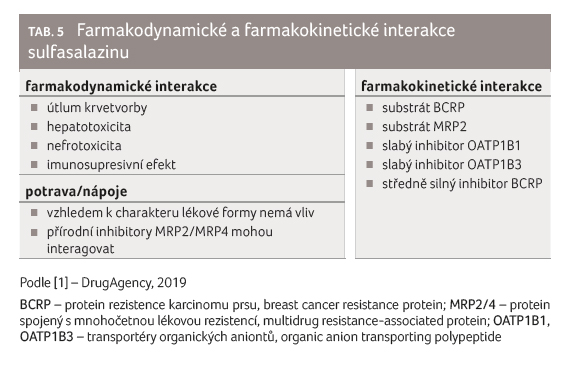
Inhibitorem BCRP je např. kurkumin, který inhibuje tento efluxní transportér již v koncentracích 1,6 μmol/l (inhibiční koncentrace ‒ IC50), což jsou koncentrace spolehlivě dosažitelné po konzumaci průměrného kari v restauraci. Ve studii u osmi zdravých dobrovolníků byla podána jednorázová dávka sulfasalazinu ve výši 0,1 mg (mikrodávka) nebo 2 g (terapeutická dávka) samotná nebo za 30 minut po předcházejícím podání kurkuminu v jednorázové dávce ve výši 2 000 mg [31]. V případě mikrodávky sulfasalazinu došlo ke zvýšení plochy pod křivkou sulfasalazinu o 83 % a ke zvýšení jeho maximálních plazmatických koncentrací o 96 %. Současně došlo ke snížení celkové clearance sulfasalazinu o 48 %. V případě terapeutické dávky sulfasalazinu došlo ke zvýšení plochy pod křivkou sulfasalazinu o 222 % a zvýšení jeho maximálních plazmatických koncentrací o 272 %. Současně došlo ke snížení celkové clearance sulfasalazinu o 69 %. Užívání kurkuminu u pacientů léčených sulfasalazinem tedy rozhodně nelze doporučit. Obdobný efekt má patrně též další polyfenol quercetin (IC50 = 0,6 μmol/l) a s velkou pravděpodobností též další polyfenoly.
Ačkoliv nebyly provedeny žádné formální studie, je vhodné, aby se pacient při terapii sulfasalazinem vyhnul současnému podávání silných inhibitorů BCRP, jako jsou lapatinib (IC50 = 0,013 μmol/l), nilotinib (IC50 = 0,025 μmol/l), regorafenib (IC50 = 0,0447 μmol/l) nebo ponatinib (IC50 = 0,013 μmol/l). Zvláštní opatrnost přitom vyžaduje případné souběžné podávání sulfasalazinu s leflunomidem, neboť leflunomid patří též k významným inhibitorům BCRP (IC50 = 0,146 μmol/l) a k takové opatrnosti přímo nabádá výrobce leflunomidu.
Sulfasalazin slabě inhibuje influxní transportní systém OATP1B1/OATP1B3, který je odpovědný za eliminaci řady léčiv, např. statinů, repaglinidu nebo některých derivátů sulfonylurey (zejména glibenklamidu). Takové případné lékové interakce obecně nejsou významné, ale mohou být v individuálních případech příčinou intolerance zmíněných léčiv.
Jak již bylo uvedeno v případě metotrexátu, je sulfasalazin inhibitorem RFC a PCFT, což má poměrně značný klinický význam, protože terapie sulfasalazinem může navodit nejen snížení vstupu metotrexátu (či leukovorinu) do buněk, ale též subklinický deficit folátu [5,6,11].
Sulfasalazin je vedle BCRP též substrátem MRP2, a proto mohou jeho biologickou dostupnost zvyšovat některá NSA [37].
Sulfasalazin může vyvolat neutropenii nebo agranulocytózu. Agranulocytóza je jedním z nejčastějších nežádoucích účinků sulfasalazinu v USA, její výskyt činí 1,60 % všech nežádoucích účinků hlášených u sulfasalazinu (n = 15 664), přičemž z 250 případů agranulocytózy bylo 234 závažných a došlo k 25 úmrtím [10]. Kombinace sulfasalazinu s léčivy, která mohou též vyvolat neutropenii či agranulocytózu, je proto nevhodná, k takovým léčivům patří tyreostatika (např. metimazol nebo propyltiouracil), analgetikum metamizol nebo většina imunosupresivně působících léčiv (včetně metotrexátu).
Sulfasalazin může též zapříčinit zhoršení funkce jater nebo ledvin a jeho kombinace s hepatotoxickými nebo nefrotoxickými léčivy nejsou vhodné.
Hydroxychlorochin
Hydroxychlorochin představuje další csDMARD používaný při terapii revmatoidní artritidy od roku 1955. Též hydroxychlorochin lze považovat za dobře účinné léčivo, které je však spojeno s relativně vysokým rizikem vzniku závažných nežádoucích účinků. Nejedná se přitom pouze o dobře známou retinopatii s defekty zorného pole, kterou jistě každý lékař pečlivě monitoruje, ale např. o prodloužení intervalu QTc nebo komorové arytmie typu torsade de pointes. Hydroxychlorochin, stejně jako ostatní deriváty 4 aminochinolinu, prodlužuje interval QTc a může tak vyvolat komorové arytmie typu torsade de pointes. V retrospektivní studii u 19 pacientů s revmatoidní artritidou nebo systémovým lupus erythematodes dlouhodobě léčených hydroxychlorochinem v dávkách 200 mg (n = 4) nebo 400 mg (n = 15) denně byla zjištěna délka QTc před zahájením terapie v průměru 424 ms (rozpětí 377‒584 ms), po 1,2‒9,2 (v průměru 3,6) roku trvající terapii hydroxychlorochinem byla délka 449 ms (rozpětí 387‒620 ms), v průměru tedy došlo k prodloužení QTc o 25 ms [38]. Podle databáze nežádoucích účinků FDA bylo k 31. 3. 2019 evidováno celkem 113 případů prodloužení intervalu QTc a 82 případů torsade de pointes, z nichž v pěti případech došlo k úmrtí pacienta [10].
Hydroxychlorochin je substrátem CYP2D6
s pravděpodobným přispěním CYP2C8 a CYP3A4 [39].
Současně je slabým inhibitorem CYP2D6 [40]. Hydroxychlorochin je
dále substrátem BCRP a nedávno bylo zjištěno, že je in
vitro poměrně silným inhibitorem OATP1A2 [41,42],
tabulka 6.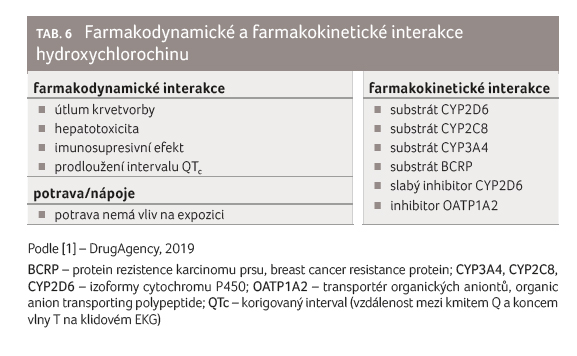
Inhibice OATP1A2 může být klinicky významná u řady léčiv. OATP1A2 je influxní transportní systém exprimovaný v tenkém střevě, který zajišťuje aktivní vstřebávání hydrofilních aniontů, jako jsou např. betablokátory (atenolol, acebutolol, celiprolol), ale také fluorovaných chinolonů (ciprofloxacin, levofloxacin) a řady dalších léčiv. Současné podávání hydroxychlorochinu by proto mohlo snížit biologickou dostupnost těchto léčiv, dosud však nebyly provedeny žádné studie.
Penicilamin
Ačkoliv je penicilamin, další csDMARD, používán při terapii revmatoidní artritidy od roku 1970, je k dispozici jen minimum informací o jeho lékových interakcích. Je známo, že se nesmí kombinovat se solemi zlata (zvýšení myelotoxicity) a s chlorochinem (zvýšení expozice penicilaminu) [43], avšak jeho kombinace s hydroxychlorochinem je možná [44].
Penicilamin je substrátem MRP2 a BCRP. Proto je možné očekávat, že NSA mohou zvýšit expozici penicilaminu, což bylo v případě indometacinu také prokázáno, když podávání indometacinu klinicky nevýznamně zvýšilo plazmatické koncentrace penicilaminu o 26 % [43].
Leflunomid
Dalším csDMARD je leflunomid, který
je používán při terapii revmatoidní artritidy od roku 1999.
Leflunomid je substrátem CYP1A2, CYP2C19 a CYP3A4. Význam
lékových interakcí v důsledku inhibice nebo indukce těchto
izoenzymů CYP450 je však patrně zanedbatelný. Leflunomid je
substrátem BCRP, OAT1/3 a MRP2 [45,46], tabulka 7.
Nejvýznamnější jsou z hlediska lékových interakcí schopnosti leflunomidu (respektive jeho metabolitu teriflunomidu) inhibovat CYP2C8 nebo BCRP. Teriflunomid (aktivní metabolit leflunomidu) zvýšil expozici substrátu CYP2C8 repaglinidu o 128 %, což je klinicky významné [47]. Teriflunomid rovněž zvyšuje expozici substrátu BCRP rosuvastatinu o 151 %, také toto zvýšení je klinicky významné, výrobce leflunomidu dokonce uvádí, že při současném podávání rosuvastatinu nesmí jeho dávka přesáhnout 10 mg denně [48]. Teriflunomid zvýšil expozici substrátu OAT3 cefakloru o 54 %. Tato léková interakce není klinicky významná, přesto výrobce leflunomidu doporučuje zvýšenou opatrnost v případě souběžného podávání se substráty OAT3, jako například s cefaklorem, benzylpenicilinem, ciprofloxacinem, indometacinem, ketoprofenem, furosemidem, cimetidinem, metotrexátem, zidovudinem.
Cyklosporin
Dalším csDMARD je cyklosporin, jehož použití nelze z důvodu převyšujících nežádoucích účinků v současné době pro léčbu revmatoidní artritidy doporučit [49]. Ve stejném duchu se k použití cyklosporinu v terapii revmatoidní artritidy vyslovují doporučené postupy [3].
Z hlediska lékových interakcí
patří cyklosporin k nejlépe prozkoumaným léčivům.
Lékových interakcí je známo velké množství a značná
část z nich je klinicky vysoce významná (tab. 8). Jedná se o lékové interakce, jejichž mechanismus je založen na inhibici transportního systému
P glykoproteinu, BCRP a OATP1B1 a OATP1B3 [50‒52].
mechanismus je založen na inhibici transportního systému
P glykoproteinu, BCRP a OATP1B1 a OATP1B3 [50‒52].
Z klinicky nejvýznamnějších lékových interakcí cyklosporinu lze za všechny uvést interakce se statiny. Podávání cyklosporinu zvyšuje expozici atorvastatinu o 769 %, fluvastatinu o 310 %, rosuvastatinu o 971 % a simvastatinu o 150 % [53‒56]. Podobně interaguje cyklosporin též s novými perorálními antikoagulancii (NOAC) a s digoxinem. Expozici cyklosporinu pak zvyšují silné (např. klaritromycin nebo azolová antimykotika) a středně silné (např. verapamil) inhibitory CYP3A4/P glykoproteinu.
Azatioprin
Ze stejných důvodů jako
v případě cyklosporinu je používání následujícího
csDMARD azatioprinu již minulostí [49]. Azatioprin je substrátem
transportního systému P glykoproteinu a tiopurin
S metyltransferáza (TPMT), jehož aktivita je polymorfní (tab. 9) [57]. Před zahájením podávání azatioprinu by měla
být vyšetřena aktivita TPMT a podle výsledku by měly být
upraveny dávky azatioprinu [58,59]. Vyšetření genetického
polymorfismu TPMT je v ČR hrazeno z prostředků veřejného
zdravotního pojištění (kód VZP ČR 94965).
Azatioprin je substrátem TPMT, inhibitorem TPMT je alopurinol. Současné podávání azatioprinu s alopurinolem lze bez snížení dávky azatioprinu považovat za zásadně nebezpečné, průměrné snížení dávky azatioprinu přitom v kontrolované studii dosahovalo 73 % [60]. Na základě publikované kazuistiky se ukazuje, že podobný, i když patrně slabší efekt jako alopurinol má i febuxostat [61]. Dalším slabším inhibitorem TPMT (ve srovnání s alopurinolem) je mesalazin a jeho prekurzor sulfasalazin. V retrospektivní studii bylo sledováno 139 pacientů s ulcerózní kolitidou nebo s Crohnovou chorobou, kteří byli léčeni alespoň 12 týdnů azatioprinem v dávkách 2 mg/kg denně (n = 132) nebo merkaptopurinem, jenž je metabolitem azatioprinu, v dávkách 1 mg/kg denně (n = 7) [62]. Celkem 94 pacientů bylo takto léčeno monoterapií azatioprinem nebo merkaptopurinem, nebo byl současně podáván též mesalazin v dávkách 3 000 mg denně. Mnohorozměrnou regresní analýzou bylo zjištěno, že při současném podávání mesalazinu bylo riziko myelotoxicity zvýšeno 3,45krát (1,31‒9,04 na 95 % hladině spolehlivosti, p = 0,01).
Lékové interakce jednotlivých
tsDMARDs
Všechna tsDMARDs jsou substráty CYP3A4. Tofacitinib je citlivým substrátem CYP3A4, a proto jeho kombinace se silnými inhibitory CYP3A4 představuje riziko dané zvýšením expozice tofacitinibu přibližně o 100 %, toto výrobce doporučuje minimalizovat redukcí dávky tofacitinibu z obvyklých 5 mg dvakrát denně na 5 mg jednou denně. V případě apremilastu a baricitinibu se však celková expozice klinicky významným způsobem nemění a silné inhibitory CYP3A4 je možné s opatrností s těmito léčivy podávat.
Odlišná je situace v případě kombinace tofacitinibu, baricitinibu a apremilastu se silnými induktory CYP3A4. Silné induktory snižují expozici tofacitinibu o 84 % a apremilastu o 72 %, z čehož výrobci vyvozují doporučení vyhnout se současnému podávání silných induktorů CYP3A4. Snížení expozice baricitinibu působením silného induktoru CYP3A4 je malé (činí 34 %) a není doprovázeno změnou plazmatických koncentrací baricitinibu, proto výrobce nedoporučuje přijetí jakýchkoliv opatření z hlediska opatrnosti nebo změn v dávkování.
V případě tofacitinibu je vhodné vyhnout se konzumaci grapefruitů nebo pití grapefruitové šťávy, protože látky v ní obsažené inhibují CYP3A4. Výrobce v ČR takové doporučení neuvádí, uvádí jej ale výrobce tofacitinibu v Kanadě. Je též vhodné vyhnout se konzumaci rostlinných přípravků obsahujících induktory CYP3A4, jako je třezalka tečkovaná (Hypericum perforatum), nebo extrakty z ní vyrobené, dále šalvěj červekořenná (Salvia miltiorrhiza), což je významná součást tradiční čínské medicíny, dále též čajovec kapský (Aspalathus linearis), známý spíše pod názvem rooibos, a nově stále více oblíbený nápoj z ostružiníku sladkého (Rubus suavissimus), což je též součást tradiční čínské medicíny a tradiční čínský nápoj.
Obdobná doporučení ohledně přírodních induktorů CYP3A4 je možné dát i pro apremilast. Naproti tomu v případě baricitinibu souběžně užívané přírodní induktory CYP3A4 patrně nevadí.
Lékové interakce jednotlivých
bDMARDs
Mohlo by se zdát, že biologická léčiva, reprezentovaná především monoklonálními protilátkami proti různým buněčným povrchovým determinantám, nemohou mít farmakokinetické lékové interakce. Jedná se totiž o proteiny, které samy o sobě nemají přímý vliv na aktivitu enzymů ani transportních systémů.
Informace o vlivu inhibitorů IL 1, IL 6 a inhibitorů TNFα na aktivitu některých enzymů byly, alespoň z experimentálních studií, dávno známé [63,64].
Donedávna nebylo známo, zda in vivo může aplikace blokátorů těchto cytokinů ovlivnit aktivitu izoenzymů CYP450. To se změnilo publikováním výsledků studie, z nichž vyplynulo, že zahájení aplikace tocilizumabu pacientům s revmatoidní artritidou klinicky významným způsobem ovlivňuje expozici substrátu CYP3A4 simvastatinu [65]. Plocha pod křivkou simvastatinu se týden po první infuzi tocilizumabu snížila o 43 % (34‒55 % na 90% hladině spolehlivosti), a pátý týden po infuzi dokonce o 61 % (47‒78 % na 90% hladině spolehlivosti). Toto snížení lze považovat za klinicky významné.
V době první publikace zmíněné studie nebylo jasné, zda stejné závěry platí i pro ostatní izoenzymy CYP450, v této době však bylo známo, že aktivita těchto izoenzymů u pacientů s chronickým zánětlivým onemocněním je nižší než u zdravých jedinců [66]. V roce 2015 pak byly publikovány obdobné výsledky pro sirukumab, z nichž vyplývá, že podávání léčiva s antagonistickým účinkem proti IL 6 potlačí sníženou expresi a navodí fyziologickou aktivitu nejen v případě CYP3A4, ale i pro CYP2C9 a CYP2C19, a trochu překvapivě zmírní expresi CYP1A2 [67]. Již jednorázová aplikace sirukumabu pacientům s revmatoidní artritidou vedla ke snížení expozice substrátu CYP3A4 midazolamu o 30‒35 %, substrátu CYP2C19 omeprazolu o 37‒45 % a substrátu CYP2C9 S warfarinu o 18‒19 % za 1, 3 a 6 týdnů po aplikaci. Naopak došlo ke zvýšení expozice substrátu CYP1A2 kofeinu o 20‒34 % rovněž za 1, 3 a 6 týdnů po aplikaci.
Jednorázová aplikace sarilumabu pacientům s revmatoidní artritidou vedla ke snížení expozice substrátu CYP3A4 simvastatinu o 54 % (42‒69 % na 90% hladině spolehlivosti) [68].
Snížení expozice substrátům CYP3A4, které bylo pozorováno pro sarilumab, tocilizumab i sirukumab, je prvním příkladem interakce nemoc‒lék‒lék, kdy k interakci dochází pouze tehdy, je li pacient úspěšně léčen (snižuje se koncentrace IL 1 nebo IL 6). Existují dokonce důkazy o korelaci závažnosti farmakokinetických změn substrátů CYP3A4, CYP2C9 a CYP2C19 v závislosti na koncentracích IL 6 [69].
Zatímco v USA se v souvislosti s informacemi o vlivu potlačení IL 1 a IL 6 na aktivitu biotransformačních enzymů začalo seriózně diskutovat o obdobném vlivu bDMARDs ze skupiny inhibitorů TNFα na aktivitu biotransformačních enzymů a řada SPC (FDA) jednotlivých přípravků již příslušnou informaci o možnosti obnovení aktivity CYP450 obsahuje (infliximab nebo adalimumab), v Evropě žádné tendence ke změně v SPC zatím nelze pozorovat.
Nelze tedy vyloučit, že infliximab, adalimumab či golimumab a případně též další inhibitory TNFα mohou působit obdobně jako inhibitory IL 6 uvedené výše.
Jak již bylo uvedeno, je třeba opatrnosti při zahajování terapie bDMARDs u pacientů, kteří užívají citlivé substráty CYP3A4, CYP2C9, CYP2C19 nebo CYP1A2 s úzkým terapeutickým oknem. K takovým léčivům patří následující substráty CYP3A4 (buspiron, everolimus, cyklosporin, midazolam, avanafil), CYP2C9 (fenytoin, warfarin), CYP2C19 (diazepam, vorikonazol) a CYP1A2 (např. tizanidin, klozapin, anagrelid). U těchto a některých dalších léčiv je třeba sledovat účinnost a výskyt nežádoucích účinků a v případě potřeby upravit jejich dávky. Takzvané rizikové období pro změnu exprese je krátké, obvykle do dvou měsíců, poté již žádné změny farmakokinetických vlastností souběžně užívaných léčiv nejsou očekávány.
Některé lékové interakce
jednotlivých NSA
Při výčtu lékových interakcí léčiv používaných v revmatologii nemůže chybět pasáž o NSA.
Všechna NSA jsou substráty CYP2C9, MRP2 a MRP4 [20]. Jak bylo uvedeno výše, jsou glukuronidy některých NSA inhibitory MRP2/MRP4, což má např. význam při eliminaci metotrexátu, který je substrátem MRP2/MRP4, podobně jako řady dalších protinádorových léčiv (paklitaxel, vinkristin). Některá NSA (např. diklofenak) působí jako inhibitory CYP2C9, což může zvyšovat riziko při souběžném podávání substrátů s úzkým terapeutickým oknem (např. warfarin, fenytoin).
Nesteroidní antirevmatika mají řadu
dalších, zejména farmakodynamických účinků, které jim
propůjčují vysoký interakční potenciál (tab. 10).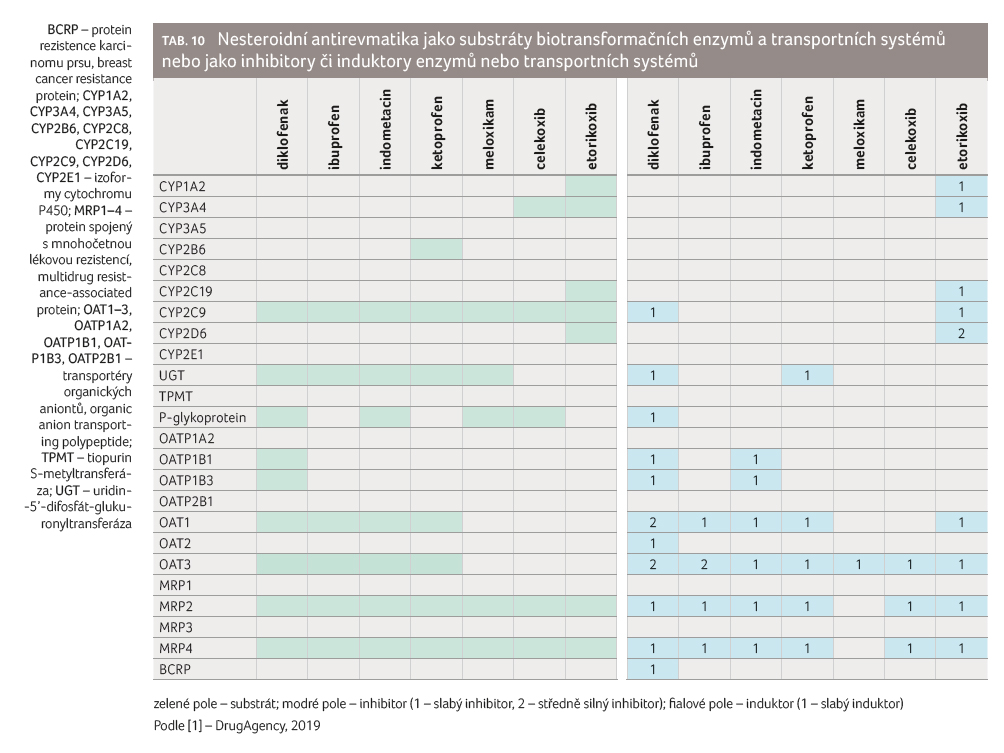
- Nesteroidní protizánětlivá léčiva
- jsou gastrotoxická (COX selektivní o něco méně) [70]
- jsou potenciálně hepatotoxická a nefrotoxická, riziko akutního selhání ledvin hrozí zejména u seniorů při kombinaci NSA, furosemid a inhibitor ACE/sartan [71‒73]
- snižují účinek antihypertenziv [74]
- zvyšují výskyt krvácivých komplikací při kombinaci s:
- perorálními antikoagulancii, včetně přímých perorálních antikoagulancií (NOAC; např. studie EINSTEIN) [75]
- antiagregancii [76]
- antidepresivy ze skupiny selektivních inhibitorů zpětného vychytávání serotoninu (SSRI) a ze skupiny inhibitorů zpětného vychytávání serotoninu a noradrenalinu (SNRI) [77,78]
- zvyšují toxicitu metotrexátu a koncentrace lithia [23,79]
- mírně zvyšují kardiovaskulární riziko (snad s výjimkou naproxenu), nikoliv však u pacientů s revmatoidní artritidou [80,81]
- zvyšují účinek perorálních antidiabetik [82]
- přispívají k hyperkalemii, především v kombinaci s dalšími léčivy jako např. inhibitory ACE [83,84]
Při souběžném podávání NSA je proto vždy třeba opatrnosti, zejména u seniorů a u pacientů souběžně léčených více léčivy.
Diskuse
Při podávání léčiv v revmatologii je nezbytné vycházet ze skutečnosti, že pacient, který trpí chronickým zánětlivým onemocněním, má odlišný metabolismus a transport léčiv ve srovnání s (jinak) zdravými jedinci. Například nemocní s revmatoidní artritidou mohou trpět celou řadou dalších onemocnění (dyslipidemií, arteriální hypertenzí, poruchou funkce jater nebo ledvin atd.). Je proto vhodné, aby se lékař při formulaci finální medikace snažil postihnout veškerou medikaci a aby byly podávány pouze léky, které jsou skutečně nezbytné.
Často lze pozorovat duplicitní podávání léčiv, kdy pacient užívá např. dva léčivé přípravky obsahující rosuvastatin nebo omeprazol. Bohužel v podmínkách ČR je tento problém poměrně významný a snad k jeho řešení přispěje pacientský lékový záznam. Nezřídka lze též pozorovat podávání léčiv, jejichž indikací si není nikdo jist a pacient je užívá mnoho let bez toho, aby byla přehodnocena potřeba takového užívání.
Z hlediska lékových interakcí se problematika může zužovat na lékové interakce založené na ovlivnění CYP450, je však nezbytné si uvědomit, že k většině farmakokinetických lékových interakcí dochází při transportu léčiv na transportních systémech, přičemž o tomto typu lékových interakcí jsou naše znalosti stále ještě velmi povrchní [85].
Zatímco v případě nově registrovaných léčiv (např. tofacitinib) jsou velmi dobře popsány jejich vlastnosti z hlediska potenciálních lékových interakcí a řada studií cílených na zjištění existence lékových interakcí je provedena a publikována, pro tzv. klasická léčiva typu hydroxychlorochin, sulfasalazin, penicilamin nebo metotrexát takovými informacemi obvykle nedisponujeme, cílené studie nebyly provedeny a publikovány a řídíme se obvykle kusými informacemi z popsaných kazuistik.
To dobře dokumentuje práce uvádějící lékové interakce uvedených klasických léčiv, kde autoři popisují asi 300 lékových interakcí, z nichž identifikovali 20 klinicky středně až velmi závažných, přičemž interagujícími léčivy byly warfarin, fluorované chinolony, azolová antimykotika, kotrimoxazol, inhibitory protonové pumpy, amiodaron, alopurinol, statiny, digoxin a hepatotoxická či nefrotoxická léčiva [86]. Často se v revmatologii zapomíná na glukokortikoidy a jejich lékové interakce.
Zajímavé stanovisko z jiného pohledu přinesla práce nizozemských autorů [87]. Autoři nejprve dle zásad evidence based medicine identifikovali důkazy pro lékové interakce v revmatologii, následně pomocí těchto informací hodnotili zjištěné konkrétní lékové interakce skupinou tří revmatologů a nezávisle na nich skupinou tří klinických farmaceutů. Obě skupiny vyhodnotily 40 potenciálních lékových interakcí, přičemž u 30 z nich se jak revmatologové, tak kliničtí farmaceuti shodli na nutnosti okamžitého zásahu. Prospektivní analýza dat ukázala, že revmatologové měli tendenci doporučit okamžitý zásah častěji, když nežádoucí reakce na lékovou interakci zahrnovala zvýšené riziko toxicity DMARDs. Naproti tomu kliničtí farmaceuti častěji obhajovali okamžitý zásah, když nežádoucí reakce zahrnovala potenciálně sníženou účinnost DMARDs. Práce jednoznačně ukazuje na výhodu spolupráce klinických lékařů s klinickými farmaceuty.
Závěr
V souvislosti s novelou zákona, který popisuje pacientský lékový záznam, se uvádělo, že v důsledku lékových interakcí zemře v ČR kolem 200 osob ročně. Databáze FAERS zaznamenává celkem 127 308 lékových interakcí, z toho 110 416 závažných, z nichž v celkem 10 508 případech došlo k úmrtí pacienta [10]. Jen za rok 2019 (leden‒červen) bylo dle FDA v USA hlášeno 7 875 lékových interakcí, z toho 7 152 závažných, z toho 777 s fatálním zakončením. Z údajů FDA vyplývá přímo exponenciální nárůst jak počtu lékových interakcí, tak počtu jejich fatálních zakončení.
Podle výskytu lékových interakcí v průběhu času jsou tyto interakce častější u žen a u pacientů vyššího věku a často bývají závažné [88,89]. Proto představují jeden z významných problémů spojených s bezpečností terapie. Lékové interakce mohou významným způsobem ovlivnit adherenci pacienta k terapii, účinnost terapie a výskyt nežádoucích účinků. Zvláštním typem lékové interakce jsou duplicity, které prakticky nikdy nepředstavují pro pacienta prospěch, ale téměř vždy pouze zvýšené riziko, nehledě na zbytečně vynaložené veřejné prostředky.
Lékové interakce tak představují nejen problém medicínský, ale potenciálně významný právní problém, který je dán případnou náhradou škody, jež může vzniknout pacientovi v důsledku nevhodné, nebo dokonce kontraindikované kombinace léčiv.
Seznam použité literatury
- [1] Databáze lékových interakcí DrugAgency, DrugAgency 2019. Dostupné na: http://lekoveinterakce.cz
- [2] Bauer S, Störmer E, Johne A, et al. Alterations in cyclosporin A pharmacokinetics and metabolism during treatment with St John’s wort in renal transplant patients. Br J Clin Pharmacol 2003; 55: 203‒211.
- [3] Šenolt L, Mann H, Závada J, Pavelka K, Vencovský J. Doporučení České revmatologické společnosti pro farmakologickou léčbu revmatoidní artritidy 2017. Dostupné na: http://www.revmatologicka‑spolecnost.cz/dokumenty/DP_RA_Senolt_2017.pdf
- [4] Graudal N, Jürgens G, Baslund B, Alderman MH. Compared with usual sodium intake, low‑ and excessive‑sodium diets are associated with increased mortality: a meta‑analysis. Am J Hypertens 2014; 27: 1129‒1137.
- [5] Jansen G, van der Heijden J, Oerlemans R, et al. Sulfasalazine is a potent inhibitor of the reduced folate carrier: implications for combination therapies with methotrexate in rheumatoid arthritis. Arthritis Rheum 2004; 50: 2130‒2139.
- [6] Urquhart BL, Gregor JC, Chande N, et al. The human proton‑coupled folate transporter (hPCFT): modulation of intestinal expression and function by drugs. Am J Physiol Gastrointest Liver Physiol 2010; 298: G248‒G254.
- [7] Weinblatt ME, Kremer JM, Coblyn JS, et al. Pharmacokinetics, safety, and efficacy of combination treatment with methotrexate and leflunomide in patients with active rheumatoid arthritis. Arthritis Rheum 1999; 42: 1322‒1328.
- [8] Wang L, Ma L, Lin Y, et al. Leflunomide Increases Hepatic Exposure to Methotrexate and Its Metabolite by Differentially Regulating Multidrug Resistance‑Associated Protein Mrp2/3/4 Transporters via Peroxisome Proliferator‑Activated Receptor α Activation. Mol Pharmacol 2018; 93: 563‒574.
- [9] Vial T, Patat AM, Boels D, et al. Adverse consequences of low‑dose methotrexate medication errors: data from French poison control and pharmacovigilance centers. Joint Bone Spine 2019; 86: 351‒355.
- [10] FDA Adverse Event Reporting System (FAERS) Public Dashboard, FDA, USA, 2019. Dostupné na: https://www.fda.gov/drugs/fda‑adverse‑event‑reporting‑system‑faers/fda‑adverse‑event‑reporting‑system‑faers‑public‑dashboard
- [11] Okada M, Fujii H, Suga Y, et al. Drug interaction between methotrexate and salazosulfapyridine in Japanese patients with rheumatoid arthritis. J Pharm Health Care Sci 2017; 3: 7.
- [12] Zamek‑Gliszczynski MJ, Zhang X, Mudunuru J, et al. Clinical Extrapolation of the Effects of Dolutegravir and Other HIV Integrase Inhibitors on Folate Transport Pathways. Drug Metab Dispos 2019; 47: 890‒898.
- [13] Pinkerton CR, Welshman SG, Glasgow JF, Bridges JM. Can food influence the absorption of methotrexate in children with acute lymphoblastic leukaemia? Lancet 1980; 2: 944‒946.
- [14] Oguey D, Kölliker F, Gerber NJ, Reichen J. Effect of food on the bioavailability of low‑dose methotrexate in patients with rheumatoid arthritis. Arthritis Rheum 1992; 35: 611‒614.
- [15] Madanat F, Awidi A, Shaheen O, et al. Effects of food and gender on the pharmacokinetics of methotrexate in children. Res Commun Chem Pathol Pharmacol 1987; 55: 279‒282.
- [16] Kozloski GD, De Vito JM, Kisicki JC, Johnson JB. The effect of food on the absorption of methotrexate sodium tablets in healthy volunteers. Arthritis Rheum 1992; 35: 761‒764.
- [17] Iwaki M, Shimada H, Irino Y, et al. Inhibition of Methotrexate Uptake via Organic Anion Transporters OAT1 and OAT3 by Glucuronides of Nonsteroidal Anti‑inflammatory Drugs. Biol Pharm Bull 2017; 40: 926‒931.
- [18] Kawase A, Yamamoto T, Egashira S, Iwaki M. Stereoselective Inhibition of Methotrexate Excretion by Glucuronides of Nonsteroidal Anti‑inflammatory Drugs via Multidrug Resistance Proteins 2 and 4. J Pharmacol Exp Ther 2016; 356: 366‒374.
- [19] Woillard JB, Debord J, Benz‑de‑Bretagne I, et al. A Time‑Dependent Model Describes Methotrexate Elimination and Supports Dynamic Modification of MRP2/ABCC2 Activity. Ther Drug Monit 2017; 39: 145‒156.
- [20] Ivanyuk A, Livio F, Biollaz J, Buclin T. Renal Drug Transporters and Drug Interactions. Clin Pharmacokinet 2017; 56: 825‒892.
- [21] Narumi K, Sato Y, Kobayashi M, et al. Effects of proton pump inhibitors and famotidine on elimination of plasma methotrexate: Evaluation of drug‑drug interactions mediated by organic anion transporter 3. Biopharm Drug Dispos 2017; 38: 501‒508.
- [22] Suzuki K, Doki K, Homma M, et al. Co‑administration of proton pump inhibitors delays elimination of plasma methotrexate in high‑dose methotrexate therapy. Br J Clin Pharmacol 2009; 67: 44‒49.
- [23] Svanström H, Lund M, Melbye M, Pasternak B. Concomitant use of low‑dose methotrexate and NSAIDs and the risk of serious adverse events among patients with rheumatoid arthritis. Pharmacoepidemiol Drug Saf 2018; 27: 885‒893.
- [24] Aherne GW, Piall E, Marks V, et al. Prolongation and enhancement of serum methotrexate concentrations by probenecid. Br Med J 1978; 1: 1097‒1099.
- [25] Lilly MB, Omura GA. Clinical pharmacology of oral intermediate‑dose methotrexate with or without probenecid. Cancer Chemother Pharmacol 1985; 15: 220‒222.
- [26] Shia CS, Juang SH, Tsai SY, et al. Interaction of rhubarb and methotrexate in rats: in vivo and ex vivo approaches. Am J Chin Med 2013; 41: 1427‒1438.
- [27] Yu CP, Hsieh YC, Shia CS, et al. Increased Systemic Exposure of Methotrexate by a Polyphenol‑Rich Herb via Modulation on Efflux Transporters Multidrug Resistance‑Associated Protein 2 and Breast Cancer Resistance Protein. J Pharm Sci 2016; 105: 343‒349.
- [28] Volk EL, Schneider E. Wild‑type breast cancer resistance protein (BCRP/ABCG2) is a methotrexate polyglutamate transporter. Cancer Res 2003; 63: 5538‒5543.
- [29] Shibayama Y, Ushinohama K, Ikeda R, et al. Effect of methotrexate treatment on expression levels of multidrug resistance protein 2, breast cancer resistance protein and organic anion transporters Oat1, Oat2 and Oat3 in rats. Cancer Sci 2006; 97: 1260‒1266.
- [30] van der Heijden JW, Oerlemans R, Tak PP, et al. Involvement of breast cancer resistance protein expression on rheumatoid arthritis synovial tissue macrophages in resistance to methotrexate and leflunomide. Arthritis Rheum 2009; 60: 669‒677.
- [31] Kusuhara H, Furuie H, Inano A, et al. Pharmacokinetic interaction study of sulphasalazine in healthy subjects and the impact of curcumin as an in vivo inhibitor of BCRP. Br J Pharmacol 2012; 166: 1793‒1803.
- [32] Ramsey LB, Mizuno T, Vinks AA, O’Brien MM. Delayed methotrexate clearance in patients with acute lymphoblastic leukemia concurrently receiving dasatinib. Pediatr Blood Cancer 2019; 66: e27618.
- [33] Wang Z, Zhang N, Chen C, et al. Influence of the OATP Polymorphism on the Population Pharmacokinetics of Methotrexate in Chinese Patients. Curr Drug Metab 2019; (v tisku) doi: 10.2174/1389200220666190701094756.
- [34] CPMP/EWP/560/95/Rev. Committee for Human Medicinal Products (CHMP): Guideline on the investigation of drug interactions. 6/2012. Dostupné na: https://www.ema.europa.eu/en/documents/scientific‑guideline/guideline‑investigation‑drug‑interactions_en.pdf
- [35] U.S. Department of Health and Human Services Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Clinical Drug Interaction Studies ‒ Study Design, Data Analysis, and Clinical Implications Guidance for Industry, 10/2017. Dostupné na: https://www.fda.gov/media/82734/download
- [36] Lee CA, O’Connor MA, Ritchie TK, et al. Breast cancer resistance protein (ABCG2) in clinical pharmacokinetics and drug interactions: practical recommendations for clinical victim and perpetrator drug‑drug interaction study design. Drug Metab Disp 2015; 43: 490‒509.
- [37] Dahan A, Amidon GL. MRP2 mediated drug‑drug interaction: indomethacin increases sulfasalazine absorption in the small intestine, potentially decreasing its colonic targeting. Int J Pharm 2010; 386: 216‒220.
- [38] Negoescu A, Thornback A, Wong E, Ostor AJ. Long QT and Hydroxychloroquine; A Poorly Recognised Problem In Rheumatology Patients. Arthritis Rheum 2013; 65(Suppl 10): 2045.
- [39] Lee JY, Vinayagamoorthy N, Han K, et al. Association of Polymorphisms of Cytochrome P450 2D6 With Blood Hydroxychloroquine Levels in Patients With Systemic Lupus Erythematosus. Arthritis Rheum 2016; 68: 184‒190.
- [40] Somer M, Kallio J, Pesonen U, et al. Influence of hydroxychloroquine on the bioavailability of oral metoprolol. Br J Clin Pharmacol 2000; 49: 549‒554.
- [41] Márki‑Zay J, Tauberné Jakab K, Szerémy P, Krajcsi P. MDR‑ABC transporters: biomarkers in rheumatoid arthritis. Clin Exp Rheumatol 2013; 31: 779‒787.
- [42] Xu C, Zhu L, Chan T, et al. Chloroquine and Hydroxychloroquine Are Novel Inhibitors of Human Organic Anion Transporting Polypeptide 1A2. J Pharm Sci 2016; 105: 884‒890.
- [43] Seideman P, Linstrom B. Pharmacokinetic interactions of pencillamine in rheumatoid arthritis. J Rheumatol 1989; 16: 473‒474.
- [44] Haagsma CJ. Clinically important drug interactions with disease‑modifying antirheumatic drugs. Drugs Aging 1998; 13: 281‒289.
- [45] Kis E, Nagy T, Jani M, et al. Leflunomide and its metabolite A771726 are high affinity substrates of BCRP: implications for drug resistance. Ann Rheum Dis 2009; 68: 1201‒1207.
- [46] Liao XY, Deng QQ, Han L, et al. Leflunomide increased the renal exposure of acyclovir by inhibiting OAT1/3 and MRP2. Acta Pharmacol Sin 2019; (v tisku) doi: 10.1038/s41401‑019‑0283.
- [47] Study 1932. In: FDA Clinical Pharmacology and Biopharmaceutics Review: Teriflunomide, Application Number 202992Orig1s000, Sanofi‑Aventis, 2012. Dostupné na: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/202992Orig1s000ClinpharmR.pdf
- [48] Weitz D, Schmider W, Menguy‑Vacheron F, et al. Teriflunomide: Potential for Transporter Mediated Drug‑Drug Interactions. Clin Pharmacol Ther 2013; 93(Suppl. 1): S112‒S113.
- [49] Zeidler HK, Kvien TK, Hannonen P, et al. Progression of joint damage in early active severe rheumatoid arthritis during 18 months of treatment: comparison of low‑dose cyclosporin and parenteral gold. Br J Rheumatol 1998; 37: 874‒882.
- [50] Dorababu M, Nishimura A, Prabha T, et al. Effect of cyclosporine on drug transport and pharmacokinetics of nifedipine. Biomed Pharmacother 2009; 63: 697‒702.
- [51] Xia CQ, Liu N, Miwa GT, Gan LS. Interactions of cyclosporin a with breast cancer resistance protein. Drug Metab Dispos 2007; 35: 576‒582.
- [52] Shitara Y, Takeuchi K, Nagamatsu Y, et al. Long‑lasting inhibitory effects of cyclosporin A, but not tacrolimus, on OATP1B1‑ and OATP1B3‑mediated uptake. Drug Metab Pharmacokinet 2012; 27: 368‒378.
- [53] Hermann M, Asberg A, Christensen H, et al. Substantially elevated levels of atorvastatin and metabolites in cyclosporine‑treated renal transplant recipients. Clin Pharmacol Ther 2004; 76: 388‒391.
- [54] Park JW, Siekmeier R, Lattke P, et al. Pharmacokinetics and pharmacodynamics of fluvastatin in heart transplant recipients taking cyclosporine A. J Cardiovasc Pharmacol Ther 2001; 6: 351‒361.
- [55] Simonson SG, Raza A, Martin PD, et al. Rosuvastatin pharmacokinetics in heart transplant recipients administered an antirejection regimen including cyclosporine. Clin Pharmacol Ther 2004; 76: 167‒177.
- [56] Arnadottir M, Eriksson LO, Thysell H, Karkas JD. Plasma concentration profiles of simvastatin 3‑hydroxy‑3‑methyl‑glutaryl‑coenzyme A reductase inhibitory activity in kidney transplant recipients with and without ciclosporin. Nephron 1993; 65: 410‒413.
- [57] Mendoza JL, Urcelay E, Lana R, et al. MDR1 polymorphisms and response to azathioprine therapy in patients with Crohn’s disease. Inflamm Bowel Dis 2007; 13: 585‒590.
- [58] Relling MV, Gardner EE, Sandborn WJ, et al. Clinical Pharmacogenetics Implementation Consortium: Clinical Pharmacogenetics Implementation Consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin Pharmacol Ther 2011; 89: 387‒391.
- [59] Relling MV, Gardner EE, Sandborn WJ, et al. Clinical Pharmacogenetics Implementation Consortium: Clinical pharmacogenetics implementation consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing: 2013 update. Clin Pharmacol Ther 2013; 93: 324‒325.
- [60] Cummins D, Sekar M, Halil O, Banner N. Myelosuppression associated with azathioprine‑allopurinol interaction after heart and lung transplantation. Transplantation 1996; 61: 1661‒1662.
- [61] Doré M, Frenette AJ, Mansour AM, et al. Febuxostat as a novel option to optimize thiopurines’ metabolism in patients with inadequate metabolite levels. Ann Pharmacother 2014; 48: 648‒651.
- [62] Gao X, Zhang FB, Ding L, et al. The potential influence of 5‑aminosalicylic acid on the induction of myelotoxicity during thiopurine therapy in inflammatory bowel disease patients. Eur J Gastroenterol Hepatol 2012; 24: 958‒964.
- [63] Chen JQ, Ström A, Gustafsson JA, Morgan ET. Suppression of the constitutive expression of cytochrome P‑450 2C11 by cytokines and interferons in primary cultures of rat hepatocytes: comparison with induction of acute‑phase genes and demonstration that CYP2C11 promoter sequences are involved in the suppressive response to interleukins 1 and 6. Mol Pharmacol 1995; 47: 940‒947.
- [64] Jäättelä M, Ilvesmäki V, Voutilainen R, et al. Tumor necrosis factor as a potent inhibitor of adrenocorticotropin‑induced cortisol production and steroidogenic P450 enzyme gene expression in cultured human fetal adrenal cells. Endocrinology 1991; 128: 623‒629.
- [65] Schmitt C, Kuhn B, Zhang X, et al. Disease‑drug‑drug interaction involving tocilizumab and simvastatin in patients with rheumatoid arthritis. Clin Pharmacol Ther 2011; 89: 735‒740.
- [66] Zanger UM, Schwab M. Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharm Ther 2013; 138: 103‒141.
- [67] Zhuang Y, de Vries DE, Xu Z, et al. Evaluation of disease‑mediated therapeutic protein‑drug interactions between an anti‑interleukin‑6 monoclonal antibody (sirukumab) and cytochrome P450 activities in a phase 1 study in patients with rheumatoid arthritis using a cocktail approach. J Clin Pharmacol 2015; 55: 1386‒1394.
- [68] Lee EB, Daskalakis N, Xu C, et al. Disease‑Drug Interaction of Sarilumab and Simvastatin in Patients with Rheumatoid Arthritis. Clin Pharmacokinet 2017; 56: 607‒615.
- [69] Jiang X, Zhuang Y, Xu Z, et al. Development of a Physiologically Based Pharmacokinetic Model to Predict Disease‑Mediated Therapeutic Protein‑Drug Interactions: Modulation of Multiple Cytochrome P450 Enzymes by Interleukin‑6. AAPS J 2016; 18: 767‒776.
- [70] Lai LH, Chan FK. Nonsteroid anti‑inflammatory drug‑induced gastroduodenal injury. Curr Opin Gastroenterol 2009; 25: 544‒548.
- [71] Meunier L, Larrey D. Recent Advances in Hepatotoxicity of Non‑Steroidal Anti‑Inflammatory Drugs. Ann Hepatol 2018; 17: 187‒191.
- [72] Harirforoosh S, Jamali F. Renal adverse effects of nonsteroidal anti‑inflammatory drugs. Expert Opin Drug Saf 2009; 8: 669‒681.
- [73] Loboz K, Shenfield GM. Drug combinations and impaired renal function ‒ the “triple whammy”. Br J Clin Pharmacol 2004; 59: 239‒243.
- [74] Ishiguro C, Fujita T, Omori T, et al. Assessing the effects of non‑steroidal anti‑inflammatory drugs on antihypertensive drug therapy using post‑marketing surveillance database. J Epidemiol 2008; 18: 119‒124.
- [75] Chan TY. Adverse interactions between warfarin and nonsteroidal antiinflammatory drugs: mechanisms, clinical significance, and avoidance. Ann Pharmacother 1995; 29: 1274‒1283.
- [76] Nam YH, Brensinger CM, Bilker WB, et al. Nonsteroidal anti‑inflammatory drug choice and adverse outcomes in clopidogrel users: A retrospective cohort study. PLoS One 2018; 13: e0193800.
- [77] Helin‑Salmivaara A, Huttunen T, Grönroos JM, et al. Risk of serious upper gastrointestinal events with concurrent use of NSAIDs and SSRIs: a case‑control study in the general population. Eur J Clin Pharmacol 2007; 63: 403‒408.
- [78] Anglin R, Yuan Y, Moayyedi P, et al. Risk of upper gastrointestinal bleeding with selective serotonin reuptake inhibitors with or without concurrent nonsteroidal anti‑inflammatory use: a systematic review and meta‑analysis. Am J Gastroenterol 2014; 109: 811‒819.
- [79] Phelan KM, Mosholder AD, Lu S. Lithium interaction with the cyclooxygenase 2 inhibitors rofecoxib and celecoxib and other nonsteroidal anti‑inflammatory drugs. J Clin Psychiatry 2003; 64: 1328‒1334.
- [80] Schjerning Olsen AM, Fosbøl EL, Gislason GH. The impact of NSAID treatment on cardiovascular risk ‒ insight from Danish observational data. Basic Clin Pharmacol Toxicol 2014; 115: 179‒184.
- [81] Bournia VK, Kitas G, Protogerou AD, Sfikakis PP. Impact of non‑steroidal anti‑inflammatory drugs on cardiovascular risk: Is it the same in osteoarthritis and rheumatoid arthritis? Mod Rheumatol 2017; 27: 559‒569.
- [82] Loza E. Systematic review on the safety of concomitant use of hypoglycemia‑inducing drugs and non‑steroidal anti‑inflammatory drugs in patients with musculoskeletal pathology. Reumatol Clin 2008; 4: 232‒239.
- [83] Lafrance JP, Miller DR. Dispensed selective and nonselective nonsteroidal anti‑inflammatory drugs and the risk of moderate to severe hyperkalemia: a nested case‑control study. Am J Kidney Dis 2012; 60: 82‒89.
- [84] Bucsa C, Moga DC, Farcas A, et al. An investigation of the concomitant use of angiotensin‑converting enzyme inhibitors, non‑steroidal anti‑inflammatory drugs and diuretics. Eur Rev Med Pharmacol Sci 2015; 19: 2938‒2944.
- [85] Cayot A, Laroche D, Disson‑Dautriche A, et al. Cytochrome P450 interactions and clinical implication in rheumatology. Clin Rheumatol 2014; 33: 1231‒1238.
- [86] Hromadkova L, Soukup T, Vlcek J. Important drug interactions in patients with rheumatic disorders: interactions of glucocorticoids, immunosuppressants and antimalarial drugs. Drugs Today (Barc) 2012; 48: 545‒553.
- [87] van Roon EN, van den Bemt PM, Jansen TL, et al. An evidence‑based assessment of the clinical significance of drug‑drug interactions between disease‑modifying antirheumatic drugs and non‑antirheumatic drugs according to rheumatologists and pharmacists. Clin Ther 2009; 31: 1737‒1746.
- [88] Guthrie B, Makubate B, Hernandez‑Santiago V, Dreischulte T. The rising tide of polypharmacy and drug‑drug interactions: population database analysis 1995‑2010. BMC Medicine 2015; 13: 74.
- [89] Češka R, Tkáč I. Určení prevalence potenciálních lékových interakcí u pacientů léčených 5 a více léčivými přípravky v České a Slovenské republice. Vnitř Lék 2016; 62: 514‒520.