Základní mechanismy transportu léčiv v organismu
Souhrn:
Pohyb léčiv v organismu a jejich koncentrace v cílovém místě jsou determinovány schopností látky přestupovat přes biologické membrány a bariéry. Přestup přes membrány zahrnuje řadu potenciálně zúčastněných mechanismů. Článek charakterizuje jednotlivé typy přestupu léčiv přes biologické membrány a bariéry a uvádí příklady léčiv, u kterých mají tyto transportní mechanismy klinický význam. Přehled shrnuje možný význam transportních mechanismů jak pro farmakokinetiku léčiv, tak pro jejich účinek a toxicitu.
Key words: drugs – transport – biological barriers – pharmacokinetics – transporters.
Summary:
The movement of drugs within the organism and their concentration in the target issue are determined by their ability to cross biological membranes and barriers. Movement across membranes may involve many different mechanisms. The article characterizes individual types of drug transport across biological membranes and barriers and offers examples of drugs in which these transport mechanisms are clinically relevant. Possible significance of transport mechanisms for both drug pharmacokinetics and effect/toxicity is also reviewed.
Obecně přijímanou skutečností je, že účinky léčiv, jak terapeutické, tak toxické, jsou závislé na koncentraci, které účinná forma dosáhne v cílovém místě. Léčivo může být na místo účinku unášeno tokem (konvekcí), což se děje např. v krvi, v intersticiálních prostorech, v cytoplazmě nebo díky Brownovu pohybu (difuzí). Ovšem při absorpci z místa podání do krve, při distribuci léčiva do buněk a do tkání či při transportu na místo eliminace musí léčivo překonat řadu bariér ve formě buněčných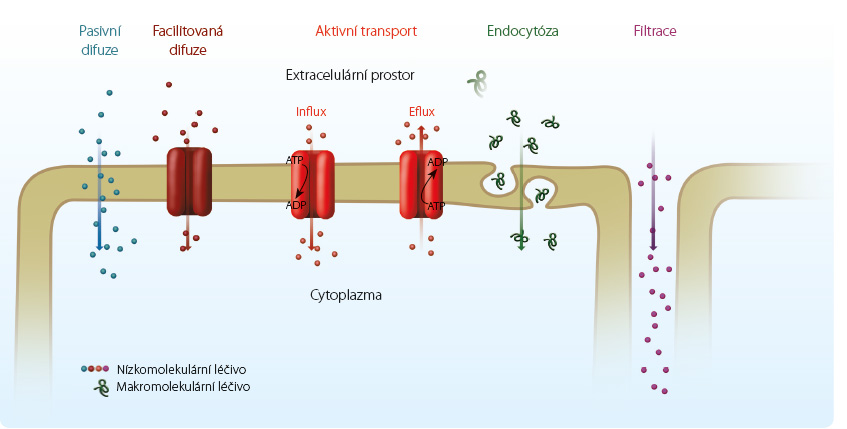 biologických membrán, které se tomuto způsobu transportu stavějí do cesty. Přestože některé látky s malou molekulou mohou přecházet přes buněčné bariéry paracelulárně nebo póry, a nemusejí tedy překračovat membrány, většiny léčiv se to netýká. Navíc v organismu je řada míst, kde mezi buňkami existují těsné spoje a paracelulární přestup není možný. Přestup přes bariéry může probíhat řadou mechanismů a je výrazně determinován chemickou podstatou léčiva [1]. Tyto transportní mechanismy lze rozdělit na pasivní difuzi, transport zprostředkovaný přenašeči (facilitovaná difuze a aktivní transport), vezikulární transport (hlavně endocytóza) a filtraci (obr. 1).
biologických membrán, které se tomuto způsobu transportu stavějí do cesty. Přestože některé látky s malou molekulou mohou přecházet přes buněčné bariéry paracelulárně nebo póry, a nemusejí tedy překračovat membrány, většiny léčiv se to netýká. Navíc v organismu je řada míst, kde mezi buňkami existují těsné spoje a paracelulární přestup není možný. Přestup přes bariéry může probíhat řadou mechanismů a je výrazně determinován chemickou podstatou léčiva [1]. Tyto transportní mechanismy lze rozdělit na pasivní difuzi, transport zprostředkovaný přenašeči (facilitovaná difuze a aktivní transport), vezikulární transport (hlavně endocytóza) a filtraci (obr. 1).
Prostá pasivní difuze
Jak již bylo řečeno, většina léčiv musí pro dosažení místa účinku překonat jednu překážku, či spíše několik bariér ve formě biologických membrán. Tyto membrány se liší obsahem lipidů, avšak všechny se vyznačují dvojvrstvou strukturou složenou především z fosfolipidů, jejichž nepolární konce jsou otočeny k sobě (dovnitř membrány) a hydrofilní hlavy tvoří vnitřní a vnější povrch membrány. Významný je obsah cholesterolu, který zvyšuje stabilitu membrán. To má význam zejména v oblastech membrán, kde mezi sebou interaguje více proteinů a plní tak signální funkce. Membrány jsou propustné pro relativně malé nepolární molekuly, přestup větších látek a molekul s nábojem je obtížný a závisí na selektivním prostupu zprostředkovaném transportními proteiny lokalizovanými v lipidové membráně.
Pasivní difuze představuje hlavní mechanismus, kterým léčiva přestupují přes biologické membrány. Jde o proces, který se uplatňuje u většiny léčiv při jejich absorpci, distribuci i eliminaci. Při pasivní difuzi se uplatňuje Fickův zákon, který říká, že přestup je závislý na schopnosti léčiva difundovat (na difuzním koeficientu), na rozdílu koncentrací na opačných stranách membrány, na velikosti plochy, přes kterou přestupují, a na tloušťce membrány. Pohyb léčiva trvá do vyrovnání koncentrací. Prakticky nejdůležitějším faktorem je, že množství přestupujícího léčiva je lineárně úměrné koncentraci na straně s vyšší hodnotou, neboť to odlišuje tento typ transportu od dalších mechanismů. Pasivní difuze není saturovatelná a je strukturně nespecifická. Aby léčivo úspěšně přestoupilo přes membránu, musí ovšem do ní nejen úspěšně vstoupit, ale na druhé straně z ní zase vystoupit. Velmi hydrofilní léčiva obtížně vstupují do membrány, zatímco příliš lipofilní látky se v membráně mohou kumulovat a z ní jen obtížně přecházet např. dovnitř buněk. Ideální pro prostup tímto mechanismem je tedy určitá vyváženost lipofility a hydrofility. Obecně lze![Obr. 2 Příklad distribuce slabé kyseliny (kyseliny acetylsalicylové, pKa 3,5) v závislosti na pH prostředí. [A‐ ] – koncentrace ionizované formy v rovnovážném stavu; [HA] – koncentrace neionizované formy v rovnovážném stavu](https://www.remedia.cz/photo-a-30867---.jpg) ovšem říci, že pasivní difuzí dobře prostupují lipofilní léčiva, hydrofilní zanedbatelně. Například rychlejší nástup centrálního účinku heroinu ve srovnání s morfinem je vysvětlován jeho vyšší lipofilitou. K popisu lipofility se u léčiv používá rozdělovací koeficient mezi oktanol a vodu (ve formě log P). Čím je log P vyšší, tím má látka vyšší lipofilitu. Vysoký log P mají látky s rychlým a vysokým průnikem do tkání – např. celková anestetika.
ovšem říci, že pasivní difuzí dobře prostupují lipofilní léčiva, hydrofilní zanedbatelně. Například rychlejší nástup centrálního účinku heroinu ve srovnání s morfinem je vysvětlován jeho vyšší lipofilitou. K popisu lipofility se u léčiv používá rozdělovací koeficient mezi oktanol a vodu (ve formě log P). Čím je log P vyšší, tím má látka vyšší lipofilitu. Vysoký log P mají látky s rychlým a vysokým průnikem do tkání – např. celková anestetika.
Pasivní difuzí mohou přestupovat i léčiva typu slabých kyselin a bází, přestože jsou relativně hydrofilní. Při každém pH existuje určitá rovnováha mezi ionizovanou a neionizovanou formou takových léčiv. Nabitá forma je hydrofilní a nepřestupuje efektivně přes biologické membrány, avšak lipofilita neionizované formy je u většiny léčiv dostatečná k přestupu pomocí pasivní difuze. Vzniká tak rovnovážný stav mezi koncentrací neionizované formy na obou stranách membrány. Protože disociace slabých kyselin a zásad je závislá na pH prostředí, které může být na opačných stranách bariéry odlišné, může být stupeň disociace a koncentrace disociované a nedisociované formy na obou stranách velmi rozdílný. Následně se postupně ustavuje rovnováha, kdy koncentrace disociovaného léčiva je i o několik řádů vyšší než na druhé straně membrány. Například v případě kyseliny acetylsalicylové bude v ideální rovnováze v relativně alkalickém prostředí plazmy s pH 7,4 díky permanentní disociaci přecházející nedisociované formy celková koncentrace přibližně 8 000× vyšší než v žaludku s pH 2,4 (obr. 2). Samozřejmě je nutné zahrnout i dynamiku změn koncentrací, které se mohou měnit v důsledku peristaltického pohybu na jedné straně a odvodu léčiva krví na druhé straně. Rovnováha bude tedy neustále narušována a přetrvávající koncentrační rozdíl ještě zvýší tendenci pohybu léčiva ze žaludku do krve. U kyselých léčiv tedy obecně platí, že se mohou částečně absorbovat už v žaludku. U bazického léčiva, jako je např. morfin, je tomu přesně naopak, neboť bude mít tendenci přecházet z krve do kyselého prostředí obsahu žaludku. Neionizovaná forma bude v kyselém prostředí vysoce ionizována, což vede k dalšímu a dalšímu přísunu neionizované formy z krve tak, aby se vyrovnaly koncentrace neionizované formy. I po parenterálním podání postupně dojde k nahromadění ionizované formy léčiva v žaludku, což je jedním z příkladů „ion trappingu“ (vychytávání iontů). Prakticky lze tento fenomén využít při otravě morfinem k rychlému odstranění významné části léčiva z těla výplachem žaludku.
Záměrnou manipulací s pH tělních tekutin lze měnit farmakokinetiku léčiv transportovaných v organismu pasivní difuzí. Příkladem je zvýšení močové exkrece slabých bází nebo kyselin úpravou pH moče. V těchto případech se významně mění disociace takových léčiv v moči a mění se míra pasivní reabsorpce nedisociované formy v ledvinných tubulech zpět do organismu. Lze podat např. bikarbonát sodný, který alkalizuje moč, a urychlit tak vylučování kyseliny acetylsalicylové při předávkování.
Transport zprostředkovaný přenašeči
Facilitovaná difuze
Na rozdíl od prosté difuze je pro přestup látek usnadněnou (facilitovanou) difuzí nutná účast integrálního membránového proteinu, který zajistí přestup přes membránu. Společným rysem s prostou difuzí je transport látek jen po koncentračním gradientu. Přenašeč (carrier) umožňuje přestup hydrofilnějš ích látek pasivním způsobem, bez vynaložení energie. U takových látek umožňuje rychlejší přestup, než se děje prostou difuzí. Dalším znakem tohoto typu transportu je saturabilita přenašeče a možnost inhibice transportu jiným substrátem či inhibitorem s afinitou k přenašeči. K tomuto typu transportu patří např. přestup zprostředkovaný transportéry skupiny OCT (transportéry organických kationtů) nebo ENT (ekvilibrační nukleosidové transportéry). Transportéry ze skupiny OCT jsou zodpovědné za uptake léčiv typu organických kationtů do hepatocytů nebo jsou součástí sekrečního systému pro tento typ léčiv v ledvinných tubulech [2]. Transportéry ze skupiny ENT přenášejí antivirové nukleosidové látky přes hematoencefalickou či placentární bariéru (tab. 1). Facilitovanou difuzí dochází také k přestupu glukózy do buněk rodinou transportérů GLUT.
ích látek pasivním způsobem, bez vynaložení energie. U takových látek umožňuje rychlejší přestup, než se děje prostou difuzí. Dalším znakem tohoto typu transportu je saturabilita přenašeče a možnost inhibice transportu jiným substrátem či inhibitorem s afinitou k přenašeči. K tomuto typu transportu patří např. přestup zprostředkovaný transportéry skupiny OCT (transportéry organických kationtů) nebo ENT (ekvilibrační nukleosidové transportéry). Transportéry ze skupiny OCT jsou zodpovědné za uptake léčiv typu organických kationtů do hepatocytů nebo jsou součástí sekrečního systému pro tento typ léčiv v ledvinných tubulech [2]. Transportéry ze skupiny ENT přenášejí antivirové nukleosidové látky přes hematoencefalickou či placentární bariéru (tab. 1). Facilitovanou difuzí dochází také k přestupu glukózy do buněk rodinou transportérů GLUT.
Aktivní transport
Podobně jako facilitovaný transport zahrnují aktivní transportní procesy účast přenašečového proteinu, který umožňuje přestup relativně hydrofilních látek přes membrány. Odlišuje se však tím, že je závislý na energii vznikající hydrolýzou adenosintrifosfátu (ATP), a tím, že transport může probíhat proti koncentračnímu spádu – z místa s menší koncentrací substrátu do míst s vyšší koncentrací. Aktivní transport je saturabilní, selektivní a může docházet ke kompetici o přenašeč mezi jednotlivými léčivy. Selektivita transportu je různá, avšak v mnoha případech je poměrně nízká, což znamená, že jeden transportér může přenášet poměrně široké spektrum léčiv – např. řadu organických aniontů či kationtů. Afinita k jednotlivým příbuzným látkám může však být velmi různá, a efektivita transportu tak velmi rozdílná. Transport může probíhat směrem do buněk – zajišťují jej influxní transportéry, nebo probíhá ven z buněk – zprostředkovávají jej efluxní transportéry. Typů transportérů, které se účastní přenosu léčiv, je velká řada. Hlavní dvě skupiny, které se uplatňují při transportu léčiv, jsou SLC (solute carrier) transportéry [2] a ABC (ATP binding cassette) transportéry. První skupina zahrnuje např. řadu transportérů pro organické anionty (OAT) či peptidy (PEPT), druhá skupina např. P glykoprotein (permeability glycoprotein) nebo BCRP (breast cancer resistance protein). Transportéry ze skupiny OAT jsou součástí sekrečního systému v ledvinách a mají význam pro močovou exkreci řady léčiv typu slabých kyselin (tab. 1). Kompetice o transportéry v ledvinách může při současném podání probenecidu zpomalit ledvinnou exkreci penicilinů, tenofoviru či inhibitorů angiotensin konvertujícího enzymu (ACE). P glykoprotein je závažnou komponentou rezistence na cytostatika ze skupin vinca alkaloidů, antracyklinů, podofylotoxinů, ale i některých nízkomolekulárních inhibitorů kináz (tab. 1), protože snižuje jejich účinné koncentrace v cílových buňkách. Činnost P glykoproteinu v membráně enterocytů je odpovědná za limitaci absorpce některých léčiv, podání inhibitorů těchto transportérů může naopak vést ke zvýšení absorpce současně podaných léčiv – substrátů P glykoproteinu. Aktivní transport se může podílet i na rezistenci na antibiotika. Příkladem je odolnost S. aureus vůči makrolidům nebo rezistence mikroorganismů k tetracyklinům.
Vezikulární transport (endocytóza a exocytóza)
Transport zprostředkovaný vezikulami zahrnuje především endocytózu – proces založený na vazbě látky na buněčnou membránu, jež se následně vychlípí a vytvoří měchýřek, který má po transportu cytoplazmou různý osud [3]. Zvláštním typem je fagocytóza, která zajišťuje příjem pevných částic do buněk. Opačný proces – výdej látek z buněk ze zde přítomných cytosolických zásobních vezikul, jejichž membrána fuzuje s buněčnou membránou a dochází k uvolnění v nich obsažené látky vně buňky – je označován jako exocytóza (obr. 3). Ta má význam především pro transport endog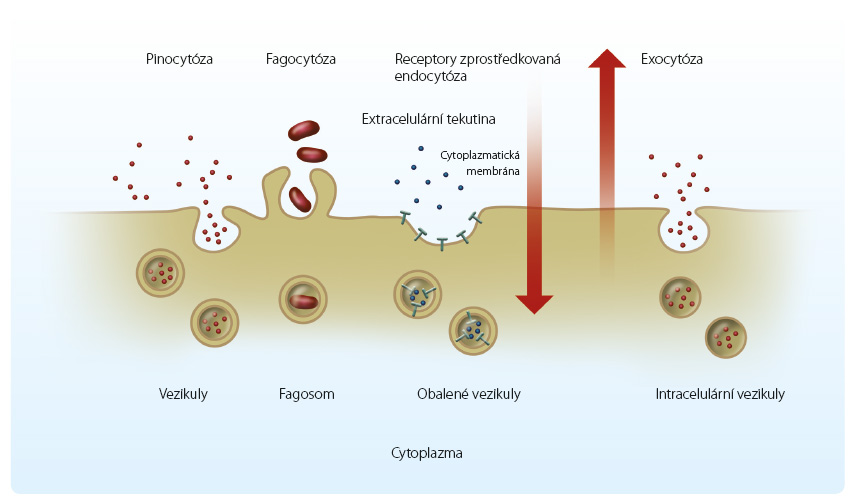 enních látek, jako je např. výdej acetylcholinu či noradrenalinu z nervových zakončení. Z hlediska transportu léčiv se jeví jako nejdůležitější endocytóza, která zahrnuje fluidní endocytózu (příjem tekutiny s obsahem léčiva) nebo endocytózu zprostředkovanou receptorem (obr. 3). Důležitým zástupcem endocytózy zprostředkované receptory je např. aktivní endocytóza zprostředkovaná systémem megalinu [4]. Tento receptor se vyskytuje v apikální membráně epiteliálních buněk ledvinných tubulů či v epitelu placenty a tenkého střeva. Megalin zprostředkovává vstup řady endogenních i exogenních proteinů, větších peptidů a některých léčiv do buněk. Nefrotoxicita aminoglykosidů je způsobena nejméně zčásti reabsorpcí a hromaděním těchto léčiv v proximálních tubulárních buňkách pomocí megalinu. Endocytózou jsou do buněk internalizovány monoklonální protilátky [5] nebo také lékové nanočástice. Transport zprostředkovaný vezikulami je intenzivně zkoumán z hlediska cíleného podávání léčiv.
enních látek, jako je např. výdej acetylcholinu či noradrenalinu z nervových zakončení. Z hlediska transportu léčiv se jeví jako nejdůležitější endocytóza, která zahrnuje fluidní endocytózu (příjem tekutiny s obsahem léčiva) nebo endocytózu zprostředkovanou receptorem (obr. 3). Důležitým zástupcem endocytózy zprostředkované receptory je např. aktivní endocytóza zprostředkovaná systémem megalinu [4]. Tento receptor se vyskytuje v apikální membráně epiteliálních buněk ledvinných tubulů či v epitelu placenty a tenkého střeva. Megalin zprostředkovává vstup řady endogenních i exogenních proteinů, větších peptidů a některých léčiv do buněk. Nefrotoxicita aminoglykosidů je způsobena nejméně zčásti reabsorpcí a hromaděním těchto léčiv v proximálních tubulárních buňkách pomocí megalinu. Endocytózou jsou do buněk internalizovány monoklonální protilátky [5] nebo také lékové nanočástice. Transport zprostředkovaný vezikulami je intenzivně zkoumán z hlediska cíleného podávání léčiv.
Filtrace
Jak už bylo naznačeno v úvodu, v organismu může docházet k přestupu léčiv přes biologické membrány či bariéry filtrací póry. Tento typ transportu je přísně limitován velikostí molekuly léčiva. Vodními póry v buněčných membránách mohou přestupovat transcelulárně jen látky do molekulové hmotnosti 100. V kapilárách však dochází díky přítomnosti velkých mezibuněčných pórů k přestupu i poměrně velkých molekul. Tento typ transportu je typický především pro ledvinné glomeruly. Glomerulární filtrace je determinována především velikostí molekuly, částečně i tvarem a nábojem filtrovaných molekul. Léčiva s molekulovou hmotností do 5 000 jsou filtrována bez omezení, se zvyšující se molekulovou hmotností se přestup přes glomerulární filtr progresivně snižuje. Za fyziologických podmínek je úplným limitem pro prostup glomerulární filtrací molekulová hmotnost přibližně 60 000–70 000. Albumin tedy za fyziologických podmínek již není vůbec filtrován. Hnací silou filtrace je hydrostatický tlak tvořený činností srdce. S poklesem filtračního tlaku se glomerulární filtrace a tím i filtrace léčiv snižuje a může dojít k jejich kumulaci v organismu a k rozvoji toxických účinků.
Závěr
Pohyb léčiva během všech farmakokinetických procesů – od absorpce po exkreci – je závislý na rychlosti a míře prostupu biologickými membránami. Tyto parametry jsou determinovány transportními mechanismy, které tento prostup zprostředkovávají. Z vlastností molekuly léčiva lze v mnoha případech odhadovat možné mechanismy transportu a chování v organismu. U mnoha léčiv je rozhodující jejich vysoká lipofilita, která umožňuje rychlý přestup přes membrány. U hydrofilních léčiv vystupuje do popředí jejich afinita k transmembránovým přenašečům. S ohledem na toxicitu a na rezistenci na léčbu je důležitá interakce léčiv s efluxními transportéry. Vzhledem k tomu, že jednotlivá léčiva mohou přecházet přes biologické membrány několika mechanismy současně, může být přesný odhad pohybu v organismu a např. místa kumulace obtížný. Na základě znalostí o transportu léčiva lze usuzovat i na toxickou potenci daného léčiva či na jeho možné interakce s jinými léčivy transportovanými stejnými systémy. Znalost způsobu transportu v organismu je tedy důležitým podkladem pro definování farmakokinetických vlastností léčiv a jejich terapeutických a jiných účinků.
Seznam použité literatury
- [1] Hacker M, Bachmann K, Messer W. (eds). Pharmacology. Principles and practice. London: Elsevier, 2009; 113–127.
- [2] Koepsell H. The SLC22 family with transporters of organic cations, anions and zwitterions. Mol Aspects Med 2013; 34: 413–435.
- [3] Doherty GJ, McMahon HT. Mechanisms of endocytosis. Annu Rev Biochem 2009; 78: 857–902.
- [4] Nielsen R, Christensen EI, Birn H. Megalin and cubilin in proximal tubule protein reabsorption: from experimental models to human disease. Kidney Int 2016; 89: 58–67.
- [5] Thurber GM, Schmidt MM, Wittrup KD. Antibody tumor penetration: transport opposed by systemic and antigen-mediated clearance. Adv Drug Deliv Rev 2008; 60: 1421–1434.