Burosumab ‒ nová naděje v léčbě hypofosfatemické křivice
Souhrn:
Kutílek Š. Burosumab – nová naděje v léčbě hypofosfatemické křivice. Remedia 2019; 29: 443–449.
Burosumab je rekombinantní lidská monoklonální protilátka IgG1, která váže fibroblastový růstový faktor 23 (FGF23) a inhibuje jeho akti-vitu. Burosumab takto zvyšuje tubulární reabsorpci fosfátů v ledvinách a koncentraci 1,25 dihydroxyvitaminu D v séru a zlepšuje minera-lizaci kostní tkáně a růst kostí. Burosumab je v současné době registrován pro léčbu X vázané hypofosfatemické rachitidy. Tento přehled uvádí základní farmakologické vlastnosti burosumabu a přináší výsledky dosavadních relevantních klinických hodnocení.
Summary:
Kutilek S. Burosumab ‒ a new chance in the treatment of hypophosphatemic rickets. Remedia 2019; 29: 443–449.
Burosumab is a recombinant human monoclonal IgG1 antibody that binds fibroblast growth factor 23 (FGF23). Burosumab increases renal tubular reabsorption of phosphate and stimulates the production of 1,25 dihydroxyvitamin D and thus improves bone mineralization and growth. Burosumab is currently marketed for the treatment of X linked hypophosphatemic rickets. This review gives basic information on burosumab pharmacology and provides results of recent relevant clinical trials.
Key words: hypophosphatemic rickets, phosphate, FGF23, burosumab
Úvod
Fosfor představuje prvek, který má klíčovou úlohu
v řadě fyziologických procesů v lidském těle. Podílí se
na struktuře a metabolismu buněk, regulaci subcelulárních procesů,
udržování acidobazické rovnováhy a je zcela nezbytný pro apoptózu hypertrofických
chondrocytů a správnou mineralizaci kostní tkáně. Fyziologické hodnoty
fosfatemie se mění s věkem (tab. 1). Homeostáza
fosforu je pro funkci organismu zásadní a podílí se na ní několik
mechanismů: střevní absorpce, renální exkrece a reabsorpce, uvolňování
fosforu z jeho zásob v kostech. Hlavními regulátory této homeostázy
jsou obsah fosforu v přijímané potravě, kalcitriol, parathormon
a fosfatoniny, z nichž nejvýznamnější je růstový faktor fibroblastů
(fibroblast growth factor 23, FGF23) [1,2].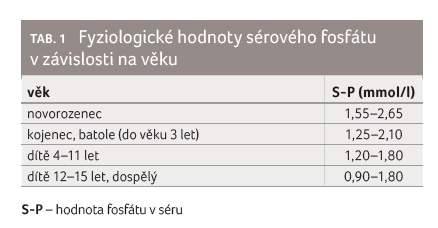
Fibroblastový růstový faktor 23 je tvořen v kostních
buňkách (osteocyty, osteoblasty), játrech, lymfatických uzlinách, thymu
a myokardu. Snižuje tvorbu jednovláknové ribonukleové kyseliny (mRNA)
pro natrium‑fosfátové kotransportéry (Npt2) v ledvinných tubulech
a tím inhibuje reabsorpci fosfátů. Dále též nepřímo ovlivňuje renální
tubulární transport fosfátů svým působením na metabolismus
vitaminu D, neboť suprimuje tvorbu enzymu 1‑alfa‑hydroxylázy
a stimuluje expresi 24‑hydroxylázy, čímž dochází k poklesu tvorby
kalcitriolu a k jeho zvýšenému katabolismu [1,2].
Proteáza PHEX (phosphate‑regulating gene with homology to
endopeptidase located on X chromosome) patří mezi povrchové membránové proteázy
závislé na zinku. Za fyziologických podmínek PHEX štěpí FGF23
a zároveň nepřímo ovlivňuje štěpení a inaktivaci FGF23
prostřednictvím enzymu konvertázy. Při inaktivační mutaci PHEX se zvyšuje
koncentrace FGF23, zvyšuje se fosfaturie, klesá tvorba kalcitriolu, což vede
k defektní mineralizaci kostní tkáně [1‒3].
Primárně hypofosfatemické křivice, jejichž hlavním
patofyziologickým podkladem jsou vysoké ztráty fosfátu močí, představují
heterogenní skupinu onemocnění. Příčinou může být zvýšená tvorba či vyšší
stabilita FGF23 nebo porušená degradace FGF23, eventuálně defektní funkce
tubulárních transportních mechanismů (tab. 2), což vede ke ztrátám
fosforu ledvinami a snížené konverzi kalcidiolu na kalcitriol [4].
Tento proces vyústí v poruchu mineralizace kostní tkáně, která se projeví
rachitidou či osteomalacií.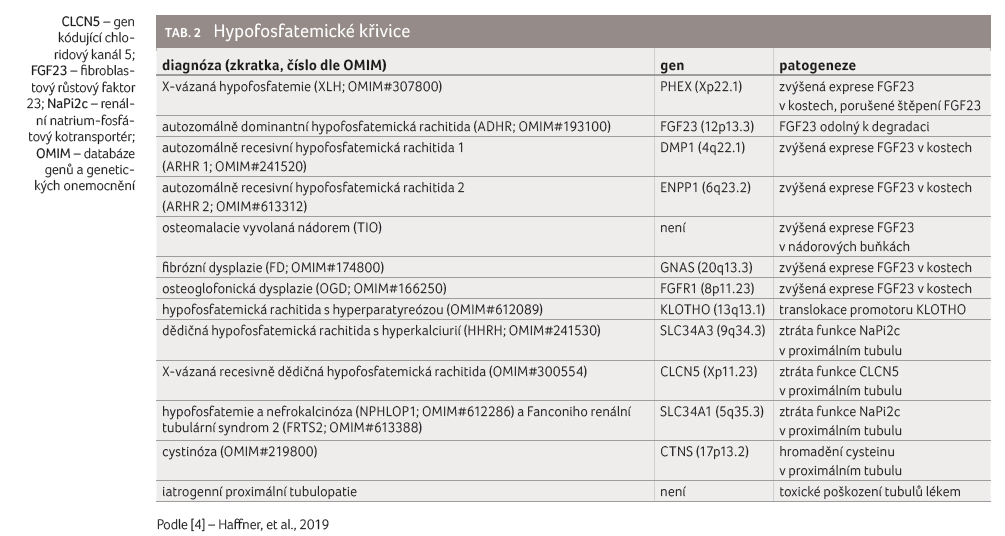
Hypofosfatemická křivice (X‑vázaná hypofosfatemie, XLH;
dříve též označována jako D‑rezistentní rachitida) je nejčastěji se
vyskytujícím zástupcem fosfopenických rachitid (incidence 1 : 20 000).
Onemocnění je charakterizované vysokými ztrátami fosforu močí, osteomalatickými
změnami až deformitami skeletu (prominující čelo, malý vzrůst, nepoměr délky
trupu a dolních končetin, genua vara, genua valga, dentální abscesy).
Příčinou XLH je inaktivační mutace PHEX. V léčbě XLH se dosud uplatňovalo
podávání fosforu a kalcitriolu (konvenční léčba). Konvenční léčba zlepšuje
tělesný růst, zmírňuje bolesti končetin, může zabránit kostním deformitám
a rozvoji dentálních abscesů, ale porucha chůze, deformity a bolesti
končetin a malý vzrůst bývají přítomny též u pacientů takto léčených.
Nežádoucími účinky konvenční léčby jsou nefrokalcinóza a sekundární až
terciární hyperparatyreóza. Jako doplňující terapie byla rovněž zkoušena
aplikace růstového hormonu ke zlepšení výsledné tělesné výšky
a podávání cinakalcetu k zamezení rozvoje sekundární a terciární
hyperparatyreózy při dlouhodobé suplementaci fosforu [5].
V současné době je převratným lékem v terapii XLH
burosumab [6].
Farmakologická skupina
Burosumab je léčivo určené k terapii nemoci kostní
tkáně pod ATC kódem M05BX05. Chemicky jde o rekombinantní lidskou
monoklonální protilátku IgG1 proti FGF23 vyráběnou metodou
rekombinantní DNA za použití savčí buněčné kultury z ovarií křečíka
čínského [7].
Indikace
Burosumab je indikován k léčbě X‑vázané hypofosfatemie
s rentgenograficky prokázanou nemocí kostí u dětí ve věku jeden
rok a starších a u dospívajících s rostoucím skeletem.
Léčbu smí zahájit pouze lékař se zkušenostmi s terapií pacientů
s metabolickými chorobami kostí.
Dávkování, způsob aplikace a monitorování léčby
Doporučená úvodní dávka je 0,4 mg/kg
tělesné hmotnosti, udržovací dávka 0,8 mg/kg
podávaná každé dva týdny. Maximální dávka je 90 mg
každé dva týdny. Všechny dávky je nutné zaokrouhlit na nejbližší desítky
miligramů. Týden před zahájením léčby je nutno ukončit podávání perorálního
fosfátu a analog vitaminu D (kalcitriol, alfakalcidiol). Koncentrace
fosfátu v séru nalačno musí být při zahájení léčby pod hranicí
referenčního rozmezí pro daný věk. Tuto hodnotu je nutné sledovat pravidelně
i po zahájení léčby. Pokud se nachází pod dolní hranicí referenčního
rozmezí pro daný věk, je možné dávku postupně zvyšovat po 0,4 mg/kg jednou za čtyři
týdny k maximální dávce. Pokud převyšuje horní hranici referenčního
rozmezí pro daný věk, musí se podávání burosumabu pozastavit a znovu
obnovit v přibližně poloviční dávce až poté, co koncentrace klesne pod
dolní hranici referenčního rozmezí.
Cílová hodnota fosfátu v séru nalačno při dolní hranici
normálního referenčního rozmezí pro daný věk snižuje riziko ektopické
mineralizace. Koncentraci fosfátu v séru nalačno je nutno stanovit
za čtyři týdny po úpravě dávky. Dávka burosumabu se nemá upravovat
častěji než jednou za čtyři týdny. Pokud pacient vynechá dávku, musí být
burosumab podán v předepsané dávce co nejdříve [7].
Burosumab je aplikován subkutánní injekcí do paže,
břicha, hýždě nebo stehna. Maximální objem léčivého přípravku na jedno
injekční místo je 1,5 ml.
Pokud je nutné v jeden den podat více než 1,5 ml, musí se celkový objem léčivého přípravku
rozdělit a aplikovat do dvou injekčních míst. Injekční místa je nutné
střídat a pečlivě sledovat s ohledem na výskyt známek možných
reakcí.
Je nutné monitorovat hodnotu fosfátu v séru nalačno
a udržovat ji při dolní hranici normálního referenčního rozmezí pro daný
věk. Dále se doporučuje sledovat každé tři měsíce koncentrace vápníku
a fosfátu v moči, každých šest měsíců koncentrace alkalické
fosfatázy, vápníku, parathormonu a kreatininu v plazmě
a na počátku léčby, každých šest měsíců a po roce léčby
jednou za rok známky a příznaky nefrokalcinózy (např.
ultrasonografickým vyšetřením ledvin).
Burosumab nebyl zkoumán u pacientů s poruchou
funkce ledvin. Burosumab se nesmí podávat pacientům s těžkým onemocněním
ledvin nebo s onemocněním ledvin v konečném stadiu.
Co se týče podávání burosumabu kojencům, bezpečnost
a účinnost léčivé látky u dětí ve věku do jednoho roku
nebyly stanoveny. Nejsou dostupné žádné údaje [7].
Zařazení do současné palety léčiv
Burosumab je určen k léčbě hypofosfatemické křivice
způsobené nadbytkem FGF23. Jedná se o biologickou léčbu XLH, která je
podstatně účinnější než dosud užívaná konvenční terapie hydroxylovaným
vitaminem D a fosfáty [7].
Farmakokinetické vlastnosti
Absorpce burosumabu ze subkutánních injekčních míst
do krevního oběhu je téměř úplná. Po subkutánním podání je doba
do dosažení maximální koncentrace burosumabu v séru (tmax) přibližně 5‒10 dnů. Maximální koncentrace v séru (cmax) a plocha pod křivkou závislosti koncentrace
burosumabu v séru na čase (AUC) jsou v rozmezí terapeuticky
podávaných dávek 0,1‒2,0 mg/kg
úměrné aplikované dávce [7‒9].
U pacientů s XLH se pozorovaný distribuční objem
burosumabu blížil objemu plazmy, což je důkazem omezené extravaskulární
distribuce [7].
Burosumab jako původní imunoglobulin je složen pouze
z aminokyselin a sacharidů a je nepravděpodobné, že by se
eliminoval prostřednictvím jaterních metabolických mechanismů. Jeho
metabolismus a eliminace probíhají cestami clearance imunoglobulinů, které
vedou k jeho degradaci na malé peptidy a jednotlivé
aminokyseliny [7].
Vzhledem k velikosti molekuly se nepředpokládá, že by
se burosumab vylučoval přímo. Clearance burosumabu závisí na tělesné
hmotnosti a odhaduje se na 0,290 litru denně u dospělé osoby
o hmotnosti 70 kg
s XLH a 0,136 l/den
u typického pediatrického pacienta (o hmotnosti 30 kg) s XLH, což odpovídá
jeho eliminačnímu poločasu (t1/2) v séru přibližně 19 dnů.
Burosumab vykazuje časově invariantní farmakokinetiku, která je lineární
vzhledem k dávce v rozmezí subkutánních dávek 0,1–2,0 mg/kg [7].
Při subkutánním podání je přímý farmakokineticko‑farmakodynamický
vztah mezi koncentracemi burosumabu v séru a zvýšenými koncentracemi
fosfátu v séru (S-P) pozorován a dobře popsán pomocí modelu
Emax/EC50. Koncentrace burosumabu a S-P, stejně jako poměr maximální
renální tubulární reabsorpce fosfátů ve vztahu ke glomerulární
filtraci (TmP/GFR), se zvyšovaly a snižovaly paralelně a dosahovaly
maximálních hodnot přibližně ve stejném časovém bodě po každé dávce,
což svědčí pro přímý farmakokineticko‑farmakodynamický vztah. Hodnota AUC pro
změnu S-P, TmP/GFR a 1,25(OH)2D se oproti začátku studie
lineárně zvyšovala se zvyšující se hodnotou AUC burosumabu [6].
Ve farmakokinetice a farmakodynamice nebyl
u pediatrických pacientů pozorován významný rozdíl v porovnání
s farmakokinetikou a farmakodynamikou v dospělé populaci.
Clearance a distribuční objem burosumabu jsou závislé na tělesné
hmotnosti [7].
Klinické zkušenosti
Farmakokinetické a farmakodynamické vlastnosti
burosumabu (původní název KRN23) podávaného v jedné dávce intravenózně či
subkutánně byly v USA hodnoceny v porovnání s placebem u 38
dospělých pacientů s XLH [7]. Podání burosumabu signifikantně zvýšilo
TmP/GFR, koncentraci S‑P a kalcitriolu (S‑1,25(OH)2D)
oproti placebu. Po subkutánním podání dosáhl S‑P maxima po 8–15 dnech
oproti intravenóznímu podání (0,5–4 dny; p < 0,01).
Změny TmP/GFR, S‑P a S‑1,25(OH)2D korelovaly
s koncentrací burosumabu v séru. Průměrný poločas burosumabu byl 8–12
dní po intravenózním podání a 13–19 dní po subkutánní
aplikaci [8].
V rámci klinické studie fáze I byly hodnoceny
farmakokinetické a farmakodynamické vlastnosti burosumabu u dospělých
pacientů s XLH, jimž bylo léčivo podáváno subkutánní injekcí každých 28
dní po dobu čtyř měsíců v postupně zvyšovaných dávkách 0,05–0,6 mg/kg a následně
po dobu 12 měsíců v titrovaných dávkách 0,1–1,0 mg/kg. Eliminační poločas
činil 17,8 dne. Doba potřebná k dosažení maximální koncentrace činila 7–10
dnů [9].
Klinické hodnocení fáze I provedené u dospělé
východoasijské populace přineslo výsledky jen nepatrně odlišné
od severoamerické studie [7,10].
Další studie byla zaměřena na hodnocení kvality života
(Quality of Life; QoL) po čtyřměsíční léčbě 28 dospělých pacientů
s XLH pomocí dotazníků SF‑36v2 Health Survey (SF‑36v2) a Western
Ontario and McMaster Osteoarthritis Index (WOMAC). Pacientům byl aplikován
burosumab subkutánně každých 28 dní. Léčba trvající čtyři měsíce výrazně
zlepšila QoL, zejména s ohledem na ústup bolesti a kloubní
ztuhlosti [11].
Vzhledem ke skutečnosti, že XLH je geneticky podmíněnou
chorobou začínající již v dětském věku a burosumab je určen převážně
dětským pacientům, byly provedeny studie u dětí s XLH.
Dvě otevřená nezaslepená hodnocení fáze II testující
burosumab celkem u 65 dětí ve věku 1–12 let se závažnou XLH
prokázala, že v krátkém období (12–16 měsíců) přinesla studijní látka
následující výsledky [7,12–14]:
- zvení TmP/GFR a následně zvýšení koncentrace S-P k dolní hranici normálního referenčního rozmezí pro daný věk se současně zvýšenými hodnotami 1,25(OH)2D;
- významné zmírnění závažnosti rachitidy na základě skóre závažnosti rachitidy (Rickets Severity Score, RSS) a radiografického globálního dojmu změny (Radiographic Global Impression of Change, RGI‑C);
-
významné zlepšení fyzických schopností (měřeno vzdáleností při šestiminutovém testu chůze, 6MWT);
-
významné zmírnění bolesti hlášené pacientem a zmírnění funkční invalidity (měřeno hodnocením Pediatrické ortopedické společnosti Severní Ameriky, Pediatric Orthopedic Society of North America Outcomes Data Collection Instrument, PODCI).
Studie UX023‑CL201
V pediatrické studii UX023‑CL201 bylo léčeno 52
pediatrických pacientů s XLH ve věku od 5 do 12 let (průměr
8,5 ± 1,87 roku [směrodatná
odchylka, SD]) po dobu 64 týdnů. U téměř všech pacientů byla
rentgenograficky prokázána přítomnost rachitidy na začátku studie
a nemocní byli dříve léčeni perorálními fosfáty a analogy vitaminu D
v průměru po dobu 7 ± 2,4 (SD) roku. Tato
konvenční terapie byla ukončena 2‒4 týdny před zahájením léčby burosumabem.
Dávka burosumabu byla upravena tak, aby se cílová koncentrace S-P nalačno
pohybovala v rozmezí 1,13‒1,62 mmol/l.
Dvaceti šesti z 52 pacientů byl burosumab podáván každé čtyři týdny,
zbývajícím 26 pacientům byl burosumab podáván každé dva týdny v průměrných
dávkách 0,73 (rozpětí 0,3‒1,5), 0,98 (0,4‒2,0) a 1,04 (0,4‒2,0) mg/kg
v 16., 40. a 60. týdnu a v maximální dávce 2,0 mg/kg [7,12].
Burosumab zvýšil koncentraci S-P a zvýšil maximální
tubulární resorpci fosfátu, TmP a TmP/GFR. Ve skupině, které byl
burosumab podáván každé dva týdny, se průměrná koncentrace S-P zvýšila
z 0,77 ± 0,13 (SD mmol/l
na začátku studie na 1,07 ± 0,13 mmol/l ve 40. týdnu a tato
koncentrace se udržela do 64. týdne s hodnotou 1,08 ± 0,14 mmol/l.
Průměrná celková aktivita alkalické fosfatázy v séru (S‑ALP)
činila 459 ± 105 (SD) U/l (7,65 ± 1,75 µkat/l) na začátku studie
a snížila se na 369 ± 76 U/l
v 64. týdnu (6,15 ± 1,27 µkat/l; rozdíl
činil 19,6 %; p < 0,0001).
Hodnoty kostní izoformy alkalické fosfatázy v séru byly
na počátku studie 165 ± 52 (SD) µg/l a 115 ± 31 µg/l
v 64. týdnu (průměrná změna ‒28,5 %)
[7,12].
Důležitým vodítkem léčby křivice je kromě aktivity S‑ALP též
skóre závažnosti rachitidy (RSS).
Toto skóre se stanovuje za použití rentgenografické
skórovací metody, která byla původně vyvinuta k posuzování závažnosti
nutriční rachitidy podle změn na zápěstích a kolenou s ohledem
na stupeň rozvláknění metafýz, jejich pohárkovitý tvar a postižení
růstové ploténky. Záporná změna skóre závažnosti rachitidy odráží zmírnění či
ústup známek rachitidy na rentgenogramu [13]. Ve studii UX023‑CL201
bylo RSS stanoveno za použití předem definované stupnice přihlížející
ke specifickým abnormalitám zápěstí a kolen. U 26 dětí, které
dostávaly burosumab každé dva týdny (viz výše), byl počáteční průměr RSS 1,92 ± 1,2 (SD), po 40 týdnech došlo k poklesu
o ‒1,06 ± 0,1 (standardní chyba, SE; p < 0,0001)
a po celkovém 64týdenním podávání burosumabu o ‒1,00 ± 0,1 (SE; p < 0,0001).
U 26 dětských pacientů, jimž byl burosumab podáván každé čtyři týdny, byl
počáteční průměr RSS 1,67 ± 1,0 (SE), po 40 týdnech
došlo k poklesu o ‒0,73 ± 0,1 (SE; p < 0,0001)
a po celkovém 64týdenním podávání burosumabu o ‒0,84 ± 0,1 (SE; p < 0,0001)
[7,12,13].
K hodnocení závažnosti rachitidy se používá též celkový
radiografický dojem změny (RGI‑C), relativní hodnoticí stupnice, která
porovnává známky rachitidy před léčbou a po léčbě. K hodnocení
se využívá sedmibodová stupnice posuzující změny stejných abnormalit jako
v případě RSS. Skóre se pohybuje od ‒3 (což představuje závažné
zhoršení rachitidy) do +3 (což představuje úplné vymizení rachitidy).
Kladná změna ve skóre RGI‑C odráží zmírnění známek rachitidy
na rentgenogramu [7,13]. U 26 dětí, které dostávaly burosumab každé
dva týdny (viz výše), byl průměr RGI‑C po 40 týdnech 1,66 ± 0,1 (SE; p < 0,0001)
a po celkovém 64týdenním podávání burosumabu 1,56 ± 0,1 (SE; p < 0,0001).
U 26 dětských pacientů, jimž byl burosumab podáván každé čtyři týdny, byl
průměr RGI‑C po 40 týdnech 1,47 ± 0,1 (SE; p < 0,0001)
a po celkovém 64týdenním podávání burosumabu 1,58 ± 0,1 (SE; p < 0,0001) [7,13].
Podávání burosumabu vedlo k významnému zlepšení
fyzických schopností (měřeno 6MWT), k významnému zmírnění bolesti hlášené
pacientem a ke zmírnění funkční invalidity (měřeno PODCI).
Interkondylární vzdálenost při genua vara a intermaleolární vzdálenost při
genua valga se od začátku studie do 64. týdne nezměnily.
V průběhu léčby se vyskytly jen tři závažné nežádoucí příhody, bez
souvislosti s terapií [7,12,13].
Studie UX023‑CL205
V pediatrické studii fáze II UX23‑CL205 byl burosumab
hodnocen u 13 pacientů s XLH ve věku 1‒4 roky (průměr 2,9 ± 1,1 roku [SD]) po dobu 64 týdnů.
U všech pacientů byla rentgenograficky prokázána přítomnost rachitidy
na začátku studie a 12 pacientů bylo dříve léčeno perorálními fosfáty
a analogy vitaminu D po dobu 16,7 ± 14,4 (SD) měsíce. Tato
konvenční terapie byla ukončena 2‒6 týdnů před zahájením léčby burosumabem.
Burosumab byl pacientům aplikován v dávce 0,8 mg/kg každé dva týdny.
Průměrná koncentrace S-P nalačno se ve studii UX023‑CL205
zvýšila z 0,81 ±
0,092 (SD) mmol/l na začátku studie na 1,12 ±
0,158 mmol/l ve
40. týdnu.
Průměrná aktivita S‑ALP činila 549 ± 193,8
(SD) U/l (9,15 ± 3,23 µkat/l)
na začátku a snížila se na 335 ± 87,6 U/l (5,58 ± 1,47 µkat/l) ve 40. týdnu studie
(průměrná změna ‒36,3 %).
Po 40 týdnech léčby burosumabem se průměrná hodnota
celkového RSS snížila z 2,92 ± 1,387 (SE) na začátku
studie na 1,19 ± 0,522 (SE; p < 0,0001)
a na 0,92 po dovršení 64 týdnů léčby.
Po 40 týdnech léčby burosumabem byla průměrná hodnota
celkového skóre RGI‑C 2,33 ± 0,08 (SE) u všech
13 pacientů (p < 0,0001) a 2,2 ± 0,1 (p < 0,0001)
po završení 64 týdnů léčby, což prokazuje zmírnění příznaků rachitidy
[6,13]. Vyskytla se pouze jedna závažná nežádoucí příhoda, bez souvislosti
s léčbou [14].
Studie UX023‑CL301
V nezaslepené multicentrické pediatrické studii fáze
III (UX023‑CL301) byl hodnocen účinek burosumabu oproti konvenční léčbě [15].
Klinického hodnocení se zúčastnilo 61 dětí s XLH, z nichž 32 bylo
randomizováno k pokračování předchozí konvenční terapie a 29
k léčbě burosumabem (0,8–1,2 mg/kg
s.c. každé dva týdny). Po 40 týdnech došlo u pacientů léčených
burosumabem k významnému zlepšení RGI‑C ve srovnání s konvenční
léčbou (p < 0,0001), rovněž tak k významnému vzestupu
hodnoty S‑P a 1,25(OH)2D, k poklesu aktivity S‑ALP,
ke zlepšení růstové rychlosti a svalové síly [15].
Další studie
U dospělých pacientů s XLH v dvojitě
zaslepeném, placebem (n = 66) kontrolovaném klinickém hodnocení fáze
III byl burosumab (n = 68) podáván subkutánně každé čtyři týdny.
Po 24 týdnech terapie došlo u skupiny léčené burosumabem
k významnému vzestupu hodnoty S‑P (p < 0,001)
a též k signifikantnímu zmírnění kloubní ztuhlosti (dotazník WOMAC; p = 0,012) [16].
V další studii, kde byly prováděny kostní biopsie,
podávání burosumabu (1,0 mg/kg
každé čtyři týdny po dobu dvou let) významně zlepšilo u dospělých
pacientů s XLH (n = 11) histomorfometrické parametry kostní
tkáně [17].
Burosumab byl registrován postupem tzv. podmíněného
schválení. Znamená to, že jsou očekávány další důkazy o jeho přínosech.
Kontraindikace a interakce s jinými
léčivými přípravky
Mezi kontraindikace podání burosumabu patří přecitlivělost
na složky přípravku, těžká porucha funkce ledvin nebo onemocnění ledvin
v konečném stadiu, koncentrace S-P nalačno nad horní hranicí normálního
rozmezí pro daný věk (z důvodu rizika rozvoje hyperfosfatemie), souběžné
podávání s perorálními fosfáty, analogy vitaminu D.
V případě kombinací burosumabu s kalcimimetiky je
nutno postupovat s opatrností. Souběžné podávání těchto léčivých přípravků
nebylo zkoumáno v klinických hodnoceních.
Nežádoucí účinky
Mezi nejčastější nežádoucí účinky patří krátkodobá lokální
reakce v místě injekce (57 %,
např. kopřivka, erytém, zduření, hematom, bolest, pruritus), bolest hlavy (54 %), bolest v končetině
(42 %), snížená koncentrace
vitaminu D (28 %),
vyrážka (23 %), bolest
zubu (19 %), zubní
absces (14 %), myalgie
(14 %) a závrať
(11 %). Nežádoucí
reakci představuje též vzestup koncentrace fosfátu v séru nalačno nad
horní hranici normálního rozmezí pro daný věk z důvodu rizika rozvoje
hyperfosfatemie a následné hyperparatyreózy [7].
Protilátky proti léku (anti‑drug antibodies, ADA) byly
detekovány u malého počtu pacientů léčených burosumabem, kteří měli
pozitivní výsledek vyšetření na přítomnost ADA také před podáním přípravku
[7].
U pacientů s XLH léčených perorálními fosfáty
a analogy vitaminu D byla pozorována ektopická mineralizace, která se
manifestovala jako nefrokalcinóza. Podávání těchto léčivých přípravků musí být
ukončeno nejméně jeden týden před zahájením léčby burosumabem. Na začátku
léčby, každých šest měsíců po dobu prvních 12 měsíců léčby a poté
jednou za rok se doporučuje sledování známek a příznaků
nefrokalcinózy, např. ultrasonografickým vyšetřením ledvin. Doporučuje se
sledování aktivity S‑ALP, kalcemie, koncentrace parathormonu a kreatininu
v plazmě každých šest měsíců (u dětí ve věku 1–2 roky každé tři
měsíce) nebo podle indikace.
Sledování hodnot vápníku a fosfátu v moči se doporučuje každé tři
měsíce [7].
Vzhledem k riziku rozvoje hyperfosfatemie je nutné
u pacienta sledovat koncentraci S-P nalačno. Za účelem snížení rizika
ektopické mineralizace se doporučuje udržovat cílovou hodnotu S-P nalačno při
dolní hranici normálního referenčního rozmezí pro daný věk. Někdy je nutné
přerušení podávání a/nebo snížení dávky. Doporučuje se pravidelné sledování
postprandiálních hodnot S-P.
U některých pacientů s XLH bylo v průběhu
léčby burosumabem pozorováno zvýšení hodnoty parathormonu v séru (rozvoj
sekundární až terciární hyperparatyreózy). Doporučuje se pravidelné sledování
koncentrace parathormonu v séru [7].
Podávání burosumabu může vyvolat lokální reakce v místě
vpichu. Podávání musí být přerušeno u všech pacientů, u nichž se
vyskytnou závažné reakce v místě vpichu, a musí být poskytnuta vhodná
lékařská terapie.
Pokud se vyskytnou závažné hypersenzitivní reakce, musí být
podávání burosumabu ukončeno a musí být zahájena vhodná terapie.
S předávkováním burosumabem nejsou zkušenosti.
Burosumab byl podáván v pediatrických klinických hodnoceních bez toxicity
limitující dávku za použití dávek až 2,0 mg/kg
tělesné hmotnosti, při maximální dávce 90 mg
každé dva týdny. V klinických hodnoceních u dospělých nebyla
pozorována toxicita limitující dávku při dávkách až 1,0 mg/kg nebo při maximální celkové dávce 128 mg každé čtyři týdny.
V případě předávkování se doporučuje přerušit podávání
burosumabu a sledovat biochemickou odpověď.
Fertilita, těhotenství a kojení
Údaje o podávání burosumabu těhotným ženám jsou omezené
nebo nejsou k dispozici. Studie na zvířatech prokázaly reprodukční
toxicitu. Podávání burosumabu se v těhotenství a u žen
v reprodukčním věku, které nepoužívají antikoncepci, nedoporučuje.
Není známo, zda se burosumab nebo jeho metabolity vylučují
do lidského mateřského mléka. Riziko pro kojené děti nelze vyloučit.
Na základě posouzení prospěšnosti kojení pro dítě a prospěšnosti
léčby pro matku je nutno rozhodnout, zda přerušit kojení, nebo ukončit/přerušit
podávání burosumabu.
Studie na zvířatech prokázaly účinky na samčí
reprodukční orgány. Klinické údaje týkající se účinků burosumabu
na fertilitu u lidí nejsou k dispozici. Zvláštní studie
fertility s burosumabem na zvířatech nebyly provedeny.
Závěr
Burosumab představuje velmi perspektivní přípravek
k dosažení úspěšné léčby hypofosfatemické křivice a osteomalacie.
Tato léčivá látka zvyšuje tubulární reabsorpci fosfátů v ledvinách
a koncentraci 1,25‑dihydroxyvitaminu D v séru, a zlepšuje tak
mineralizaci kostní tkáně a růst kostí. Terapie burosumabem prokazuje
větší účinnost než dosud užívaná konvenční léčba hydroxylovaným vitaminem D
a fosfáty.
Seznam použité literatury
- [1] Rowe PS. The wrickkened pathways of FGF23, MEPE and PHEX. Crit Rev Oral Biol Med 2004; 15: 264‒281.
- [2] Rowe PS, Garrett IR, Schwarz PM, et al. Surface plasmon resonance confirms that MEPE binds to PHEX via the MEPE ASARM motif: a model for impaired mineralization in X linked rickets (HYP). Bone 2005; 36: 33‒46.
- [3] Brewer JR, Mazot P, Soriano P. Genetic insights into the mechanisms of FGF signaling. Genes Dev 2016; 30: 751–771.
- [4] Haffner D, Emma F, Eastwood DM, et al. Clinical practice recommendations for the diagnosis and management of X linked hypophosphataemia. Nat Rev Nephrol 2019; 15: 435‒455.
- [5] Skrinar A, Dvorak Ewell M, Evins A, et al. The lifelong impact of X Linked Hypophosphatemia: Results from a burden of disease survey. J Endocr Soc 2019; 3: 1321‒1334.
- [6] Collins M. Burosumab: At long last, an effective treatment for FGF23 Associated Hypophosphatemia. J Bone Miner Res 2018; 33: 1381‒1382.
- [7] European Medicines Agency, 2018. Crysvita. Annex I — summary of product characteristics. Dostupné na: http://www.ema.europa.eu/en/documents/product information/crysvita epar product information_en.pdf
- [8] Carpenter TO, Imel EA, Ruppe MD, et al. Randomized trial of the anti FGF23 antibody KRN23 in X linked hypophosphatemia. J Clin Invest 2014; 124: 1587–1597.
- [9] Zhang X, Peyret T, Gosselin NH, et al. Population pharmacokinetic and pharmacodynamic analyses from a 4 month intradose escalation and its subsequent 12 month dose titration studies for a human monoclonal anti FGF23 antibody (KRN23) in adults with X linked hypophosphatemia. J Clin Pharmacol 2016; 56: 429‒438.
- [10] Cheong HI, Yoo HW, Adachi M, et al. First in Asian phase I study of the anti fibroblast growth factor 23 monoclonal antibody, burosumab: Safety and pharmacodynamics in adults with X linked hypophosphatemia. JBMR Plus 2018; 3: e10074.
- [11] Ruppe MD, Zhang X, Imel EA, et al. Effect of four monthly doses of a human monoclonal anti FGF23 antibody (KRN23) on quality of life in X linked hypophosphatemia. Bone Rep 2016; 5: 158‒162.
- [12] Carpenter TO, Whyte MP, Imel EA, et al. Burosumab Therapy in Children with X Linked Hypophosphatemia. N Engl J Med 2018; 378: 1987‒1998.
- [13] Thacher TD, Pettifor JM, Tebben PJ, et al. Rickets severity predicts clinical outcomes in children with X linked hypophosphatemia: Utility of the radiographic Rickets Severity Score. Bone 2019; 122: 76‒81.
- [14] Whyte MP, Carpenter TO, Gottesman GS, et al. Efficacy and safety of burosumab in children aged 1 4 years with X linked hypophosphataemia: a multicentre, open label, phase 2 trial. Lancet Diabetes Endocrinol 2019; 7: 189‒199.
- [15] Imel EA, Glorieux FH, Whyte MP, et al. Burosumab versus conventional therapy in children with X linked hypophosphataemia: a randomised, active controlled, open label, phase 3 trial. Lancet 2019; 393: 2416‒2427.
- [16] Insogna KL, Briot K, Imel EA, et al. A Randomized, Double Blind, Placebo Controlled, Phase 3 Trial Evaluating the Efficacy of Burosumab, an Anti FGF23 Antibody, in Adults With X Linked Hypophosphatemia: Week 24 Primary Analysis. J Bone Miner Res 2018; 33: 1383‒1393.
- [17] Insogna KL, Rauch F, Kamenický P, et al. Burosumab improved histomorphometric measures of osteomalacia in Adults with X Linked Hypophosphatemia: A Phase 3, Sin-gle Arm, International Trial. J Bone Miner Res 2019 Aug 1; doi: 10.1002/jbmr.3843. [Epub ahead of print].