Onasemnogen abeparvovek
Souhrn:
Haberlová J, Horák P. Onasemnogen abeparvovek. Remedia 2021; 31: 450–454.
Onasemnogen abeparvovek představuje genovou terapii založenou na adeno‑asociovaném virovém vektoru v intravenózní infuzi nesoucí transgen kódující lidský protein pro přežití motoneuronu. Jedná se o první příklad registrované systémové genové léčby v medicíně. V Evropské unii je onasemnogen abeparvovek registrován od května 2020 a je indikován k léčbě omezené skupiny pacientů s diagnózou spinální svalové atrofie – vzácného a bez dostupnosti kauzální léčby ve většině případů fatálního onemocnění. Efekt této jednorázové léčby je prokázán jak v klinických studiích, tak daty z reálného života. Nejvýraznější účinnost léčby je zaznamenána u presymptomatických pacientů. Úskalím uvedené terapie je profil nežádoucích účinků, jež mohou být ve zcela výjimečných případech velmi závažné.
Summary:
Haberlova J, Horak P. Onasemnogene abeparvovec. Remedia 2021; 31: 450–454.
Onasemnogene abeparvovec (OA) is a gene therapy based on the adeno‑associated viral vector intravenous infusion carrying a transgene to synthesize survival motor neuron protein. It is the first example of a registered systemic gene therapy in medicine. OA was registered in the EU in May 2020 and is indicated for a subgroup of patients with a diagnosis of spinal muscular atrophy (SMA). SMA is a rare, in most cases fatal disease without causal treatment. The effectiveness of this one‑time therapy has been proven with clinical trials as well as real world data. Presymptomatic therapy has the most significant efficacy. The difficulty is the profile of side effects that can be extremely severe in exceptional cases.
Key words: onasemnogene abeparvovec, treatment of spinal muscular atrophy – treatment efficacy – side effects.
Úvod
Onasemnogen abeparvovek (OA) představuje genovou terapii založenou na adeno asociovaném virovém vektoru v intravenózní infuzi. Jde o rekombinantní self komplementární adeno asociovaný virus sérotypu 9 (AAV9) obsahující transgen kódující lidský protein pro přežití motoneuronu (survival motor neuron, SMN) pod kontrolou hybridního promotoru složeného z cytomegalovirového enhanceru a kuřecího β aktinového promotoru. OA (pod obchodním názvem Zolgensma; ATC kód M09AX09) je indikován pro léčbu pacientů s diagnózou spinální svalové atrofie (spinal muscular atrophy, SMA).
Základní charakteristika spinální svalové atrofie
Spinální svalová atrofie je geneticky podmíněné onemocnění, jež je ve většině případů (95 %) vázáno na gen SMN1. Mutace v SMN1 snižuje koncentraci proteinu SMN, který je kritický k přežití motoneuronů předních rohů míšních. Při jeho absenci dochází k postupné degeneraci motoneuronů předních rohů míšních a sekundárně k poškození kosterních svalů, které je nevratné a klinicky se projeví progredující svalovou slabostí. Co do tíže klinických obtíží je SMA variabilní onemocnění. Prozatím hlavním faktorem ovlivňujícím tíži fenotypu je počet kopií genu SMN2 [1]. Klinicky nejtěžší formy SMA (typ 1 a 2) se projevují již v časném kojeneckém věku, tito pacienti mívají do tří kopií SMN2. Uvedené dvě formy tvoří většinu (až 85 %) všech pacientů trpících SMA. Pokud bychom tyto pacienty neléčili, nikdy by nebyli schopni samostatné chůze a u všech by postupně došlo k rozvoji dechové nedostatečnosti a k předčasnému úmrtí. V dobách nedostupnosti kauzální léčby byla SMA nejčastější příčinou úmrtí na geneticky podmíněné onemocnění v kojeneckém věku. Jedná se o vzácné onemocnění, incidence dle literatury je udávána 1 : 10 000 nově narozených dětí, tzn. každý rok se v ČR narodí okolo 11 pacientů s vlohou pro SMA [2]. SMA má autozomálně recesivní typ dědičnosti. Ve většině geneticky potvrzených případů jsou rodiče tzv. přenašeči nemoci, klinicky jsou zcela asymptomatičtí. Prevalence přenašečů je dle literatury u kavkazské populace 1 : 38 [3].
Princip léčby
Mechanismem účinku OA je přenos
syntetické humánní komplementární DNA (cDNA) genu SMN1
do těla pacienta pomocí virového vektoru. Jako virový vektor
je zde použit AAV9, který má vysokou afinitu k nervovým
buňkám, prochází hematoencefalickou bariérou. Cesta podání je
nitrožilní, jednorázová hodinová infuze vpraví do těla
pacienta 1,1 1014
virového genomu (vg)/kg. Virový vektor umožní vazbu na cílové
buňky, motoneurony předních rohů míšních, a následně
uvolněná kapsida se syntetickou cDNA prochází cytoplazmou
do jádra buňky, kde zůstává uložena izolovaně mimo DNA
pacienta, v tzv. epizomu. V dělících se buňkách se
syntetická DNA proto nepřenáší do dceřiných buněk.
Jelikož motoneurony předních rohů míšních nejsou schopny
replikace, předpokládá se dlouhodobý efekt léčby. Prozatím
jsou u OA dostupná data o šestiletém efektu [4].
Syntetická cDNA SMN1 je pod kontrolou hybridního promotoru
složeného z cytomegalovirového enhanceru a kuřecího
β aktinového promotoru, což mimo jiné zaručuje rychlou
transkripci, a tedy tvorbu proteinu SMN (obr. 1) [5].
Jednorázová infuze OA tak v podstatě vyřeší příčinu
nemoci, nahradí chybějící gen genem syntetickým. OA je prvním
příkladem registrované systémové genové léčby v medicíně
vůbec.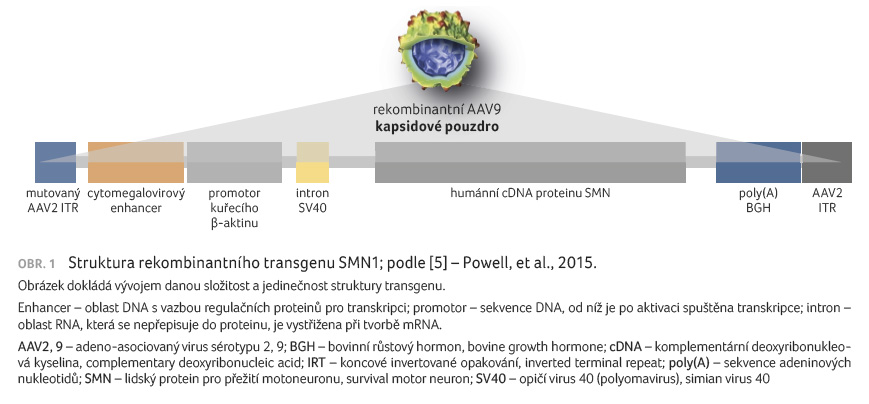
Příprava a podání onasemnogen abeparvoveku
Léčivý přípravek Zolgensma je k dispozici v lékové formě infuzního roztoku (nominální koncentrace 2 1013 vg na ml). Dostupný je ve dvou typech lahviček s (extrahovatelným) objemem 5,5 ml a 8,3 ml. Dávkování je dle hmotnosti pacienta (1,1*1014 vg/kg), dostupné je doporučení pro hmotnost od 2,6 kg do 21 kg. Lék je individuálně vyráběn pro daného pacienta mimo ČR (podle jeho hmotnosti finální balení obsahuje potřebnou kombinaci 5,5ml a 8,3ml lahviček). Aktuálně je do ČR dovážen letecky z USA zmražený na –60 °C, při transportu je lék chráněn před světlem. Po doručení do lékárny centra je OA třeba za kontrolovaných podmínek rozmrazit (podle velikosti balení trvá rozmrazení v chladničce 12-16 hodin), uchovávat v chladničce při teplotě 2-8 °C a pro ochranu před světlem ponechat v původním kartonovém obalu. Po doručení do lékárny je nutné OA spotřebovat co nejdříve, nejpozději však do 14 dnů. Alternativním postupem je rozmrazení na pokojovou teplotu těsně před podáním (podle velikosti balení trvá 4-6 hodin). Rozmrazené lahvičky není možné znovu zmrazit.
Infuzi je nutné připravit asepticky za sterilních podmínek a dodržení ochrany připravujícího personálu, ve FN Motol se prakticky používají izolátory. Celý objem odpovídající dávce pro konkrétního pacienta je asepticky přenesen do jedné až dvou 50ml injekčních stříkaček typu luer lock (závitových). Před adjustací a po ní je třeba roztok vizuálně zkontrolovat, před odběrem objemu z lahvičky obsah promísit jemným kroužením, s lahvičkou se nesmí třepat. Nejpozději do osmi hodin po adjustaci dávky do stříkačky musí být OA plně podán pacientovi. Stříkačka s dávkou OA je naplněna v lékárně a dopravena na místo podání v obalu nebo boxu pro přenos nebezpečných biologických látek (biohazard). OA je podán během hodinové infuze do periferní žíly pumpou. Nejsou k dispozici žádné údaje o kompatibilitě s jinými léčivými přípravky, OA se tedy nesmí s jinými léčivými přípravky mísit.
Jelikož se jedná o léčivý přípravek patřící do skupiny geneticky modifikovaných léčiv, je nutné při podání i během celé hospitalizace dodržovat odpovídající hygienická a režimová opatření, zejména hygienu rukou. První měsíc po podání je stolice vždy balena do dvojitého obalu a zlikvidována v rámci běžného domácího odpadu.
Farmakokinetika
OA se většinově vylučuje stolicí v průběhu prvních 30 dnů od podání. První den po podání byl OA v 0,01-0,1 % detekován taktéž ve slinách a moči. U dvou případů předčasného úmrtí, které nemělo příčinnou souvislost s podáním léku, byl OA 1,7 měsíce a 5,7 měsíce po podání detekován nejvíce v játrech, dále ve většině tkání těla (ve slezině, srdci, pankreatu, ledvinách, v tříselných lymfatických uzlinách, kosterním svalu, periferních nervech, plicích, thymu, střevech), ale i v mozku a motoneuronech předních rohů míšních, kde byla prokázána rovněž exprese proteinu SMN [6].
Preklinická data o toxicitě na zvířecích modelech
V této oblasti je nutné zmínit studii na mladých dospělých subhumánních primátech, kdy po podání OA v dávce 3 × 1013 vg/kg intratekálně byl pozorován monocytární zánět v oblasti dorzálních ganglií s důsledkem vzniku nekrózy. Relevance těchto dat pro klinickou praxi je prozatím nejasná [6].
Lékové interakce
Do dnešní doby není známa žádná interakce s jinými léčivými přípravky, je však doporučeno upravit očkovací schéma, viz kapitolu Příprava pacienta k podání léku [7].
Indikace
První zemí, kde byl OA registrován, byly Spojené státy americké. Registraci OA získal již v květnu 2019 s omezením pro pacienty ve věku maximálně dvou let trpící SMA podmíněnou bialelickou patogenní variantou v SMN1 s vazbou na chromozom 5q [8].
OA je registrován v Evropské unii od května 2020, v roce 2015 získal statut léčiva na vzácná onemocnění (tzv. orphan drug) [7]. Indikováni jsou všichni pacienti s diagnózou SMA podmíněné bialelickou patogenní variantou v SMN1 s vazbou na chromozom 5q a s fenotypem SMA typu 1 či fenotypem s maximálním počtem do tří kopií SMN2. Jedná se o tzv. podmínečné schválení (conditional marketing authorization). Indikační kritéria Evropské lékové agentury (EMA) nejsou jednotlivými evropskými zeměmi plně naplňována, v různých regionech existují různé podmínky úhrady. V České republice aktuálně platí podmínky úhrady z července 2020, kdy je lék hrazen všem pacientům s diagnózou SMA podmíněné bialelickou patogenní variantou v SMN1, kteří jsou ve věku do dvou let (definováno jako den před 3. narozeninami) a mají tělesnou hmotnost nižší než 13,5 kg či rovnou této hodnotě. Další nutnou podmínkou úhrady je celkový pacientův interní stav a neurologický nález, který nenavyšuje riziko výskytu nežádoucích účinků natolik, aby převážilo možný prospěch z léčby [9]. Tyto podmínky plně odpovídají evropskému doporučení dětských neurologů z loňského roku [10]. Indikační kritéria v ČR se do budoucna mohou měnit, závisejí zejména na dostupnosti dat léčených pacientů z jiných věkových i hmotnostních kategorií – podrobně viz kapitoly o klinických studiích a datech z praxe.
Širší indikační omezení jsou dle schváleného Souhrnu údajů o přípravku platná pro následující kategorie: pacienti s poruchou funkce ledvin či jater, nezralí novorozenci a novorozenci s hmotností nižší než 2,6 kg, pacienti s hmotností vyšší než 13,5 kg. U těchto skupin pacientů není dostatek dostupných dat o bezpečnosti či efektivitě léčby [7].
Léčba OA je centrová a je poskytována ve čtyřech neuromuskulárních centrech v ČR, kontakt na daná centra lze nalézt na stránkách Společnosti dětské neurologie ČLS JEP –www.detskaneurologie.cz.
Nežádoucí účinky
V současnosti není známa žádná lidská nemoc způsobená AAV9. Přesto má léčba OA zásadní, ve zcela výjimečných případech i fatální nežádoucí účinky. Do dnešní doby bylo OA léčeno tisíc pacientů a jsou popsány čtyři závažné skupiny nežádoucích účinků: trombocytopenie s maximem výskytu týden po podání (4 % pacientů); rozvoj trombotické mikroangiopatie (nově popsán až po registraci léku) s následným poškozením ledvin s maximem výskytu 1.-2. týden po podání; hepatopatie (12 % pacientů) se dvěma maximy - během prvního měsíce a významnější hepatopatie pak ve druhém měsíci po podání; vzestup koncentrace troponinu 1 popisovaný zejména v prvním měsíci po podání (3 % pacientů). Častými, klinicky méně závažnými nežádoucími účinky jsou opakovaný vomitus (u 8 % pacientů) v průběhu prvního a druhého týdne po podání a febrilie s maximem výskytu druhý až pátý den po podání [6]. Jednoznačný patomechanismus nežádoucích účinků znám není, přesto dle prozatím publikovaných dat se nejspíše jedná o akutní imunitní reakci zprostředkovanou zejména T lymfocyty. O příčině vomitu lze spekulovat, vektor AAV9 je prokazatelný ve stolici pacientů v průběhu celého prvního měsíce po podání. Lze předpokládat, že spektrum zatím publikovaných nežádoucích účinků se vzhledem ke krátké klinické zkušenosti bude ještě rozšiřovat. Je proto zásadní řádná evidence všech nežádoucích účinků.
Závažné nežádoucí účinky
Do současnosti byly dle znalostí autorů popsány případy akutního jaterního selhání s klinickým obrazem ikteru, koagulopatie a encefalopatie s úpravou po léčbě kortikoidy [6].
Fatální nežádoucí účinky
Informace o fatálních nežádoucích účincích pocházejí z léčby prováděné mimo klinické studie. Prozatím jsou dle znalostí autorů popsány jednotlivé případy akutního selhání ledvin jako důsledek trombotické mikroangiopatie [6].
Prevence rozvoje nežádoucích účinků
Z výše uvedených důvodů je jako prevence rozvoje nežádoucích účinků u všech pacientů doporučena imunosuprese prednisonem. Preventivní dávka prednisonu činí 1 mg/kg/den, podávání je zahájeno den před aplikací infuze a ponechává se minimálně měsíc po podání v plné dávce, následně se postupně během 28 dnů podávání ukončuje. Pokud během prvního měsíce nedojde k poklesu hodnot transamináz pod dvojnásobek normy, prodlužuje se léčba prednisonem vždy do další plánované kontroly [7]. V případě závažné hepatopatie je nyní nově německými autory doporučeno v případě zvýšení hodnot transamináz nad 6,67 μmol/l navýšit dávku prednisonu na 2 mg/kg/den po dobu minimálně jednoho týdne a v případě růstu hodnot transamináz nad 16,67 μmol/l podávat metylprednison v dávce 20 mg/kg/den po dobu tří dnů (probíhající evropská diskuse).
Příprava pacienta k podání léku
Podstatným faktorem minimalizace nežádoucích účinků je řádná příprava pacienta k podání.
Nepodkročitelnou podmínkou podání je negativita protilátek proti AAV9. Standardně tento test není ve většině zemí Evropy dostupný, proto farmaceutická společnost zajišťuje testování v evropské akreditované laboratoři v Rotterdamu. Jako negativní test je definován titr protilátek nižší než 1 : 50. Vzorek séra pacienta je transportován do laboratoře a výsledek je následně dostupný během několika dnů (z osobní zkušenosti po dohodě s laboratoří i za dva pracovní dny).
Jelikož jsou nežádoucí účinky způsobeny imunitní reakcí, nedoporučuje se léčbu podávat u pacientů s akutní či chronicky probíhající infekcí. Dále je doporučena úprava očkovacího schématu před podáním a následně po dobu užívání imunosuprese, kdy jsou zcela kontraindikovány živé vakcíny. V rizikových měsících je doporučeno před léčbou podat preventivní pasivní imunizaci proti respiračnímu syncytiálnímu viru.
Před podáním je nutné u pacientů provést základní laboratorní testy zahrnující kompletní krevní obraz včetně stanovení hodnot trombocytů a hemoglobinu, stanovení koncentrace troponinu 1, alaninaminotransferázy, aspartátaminotransferázy, celkového bilirubinu a kreatininu [7]. Ostatní vstupní testy nejsou povinné, probíhá však mezinárodní diskuse o potřebě jejich rozšíření, například o smysluplnosti stanovení hodnot protilátek proti hepatotropním virům, vyšetření renálních funkcí či rozšířeného panelu poruch koagulace.
Dispenzarizace pacienta po podání léčby
Jak již bylo řečeno, možné nežádoucí účinky mohou být závažné až fatální, je proto nutné monitorovat pacienta po podání léku minimálně po dobu tří měsíců. Pravidelné kontroly krevního obrazu a jaterních testů jsou v prvním měsíci doporučeny každý týden, ve druhém a třetím měsíci pak každé dva týdny. V případech abnormálních nálezů jsou kontroly doporučeny častěji, vždy po dohodě s danými specialisty [7].
Těhotenství a kojení
Do dnešní doby nejsou známa žádná data o léčených pacientech v průběhu těhotenství či laktace. Pokud je autorům známo, nebyly provedeny žádné zvířecí klinické studie hodnotící plodnost a fertilitu po podání OA [7].
Dostupná data z klinických studií
Klinická studie AVXS 101 CL 303 představuje otevřené multicentrické hodnocení fáze III s OA v dávce 1,1 1014 vg/kg. Ve studii bylo léčeno 22 pacientů se SMA typu 1 a 2 s kopiemi SMN2 ve věku do šesti měsíců a s hmotností do 7,5 kg. Jako primární parametr účinnosti byl hodnocen stav ventilace u pacientů ve věku 14 měsíců, kdy v 90,9 % byla zachována spontánní ventilace a tito nemocní nepotřebovali dechovou podporu. Uvedená data jsou v rozporu s přirozeným průběhem nemoci, kdy až 40 % pacientů se SMA typu 1 umírá ve věku do jednoho roku. Sekundárním parametrem posuzování účinnosti byla motorická dovednost pacientů hodnocená pomocí funkční škály pro vyšetření neuromuskulárních poruch u dětí CHOP INTEND [11]. Ve studii bylo 14 z 22 pacientů ve věku 14 měsíců schopno samostatného sedu na dobu delší než 30 sekund. Toto zjištění je opět zcela v rozporu s přirozeným průběhem nemoci, kdy pacienti se SMA typu 1 nejsou nikdy schopni samostatného sedu [12,13].
Klinická studie AVXS 101 CL 101 byla otevřeným hodnocením fáze I jednoho centra, které sledovalo podání OA v dávce 1,1 1014 vg/kg. Ve studii bylo léčeno 12 pacientů se SMA typu 1 a 2 s kopiemi SMN2 ve věku do osmi měsíců a o hmotnosti do 8,5 kg. Ve věku 14 měsíců žádný z léčených pacientů nevyžadoval umělou plicní ventilaci a ve věku 24 měsíců 9 z 12 pacientů bylo schopno samostatného sedu po dobu více než 30 sekund [14].
Klinická studie AVXS 101 CL 304 je stále probíhajícím otevřeným multicentrickým hodnocením s presymptomatickými pacienty se SMA ve věku do šestého týdne života a se dvěma (soubor 14 nemocných) či třemi kopiemi SMN2 (soubor 15 nemocných). V době hodnocení studie (12/2019) byli všichni pacienti v obou skupinách bez nutnosti umělé plicní ventilace. Sedm z osmi pacientů ve skupině 1 dosáhlo samostatného sedu ve věku odpovídajícím věku zdravého dítěte a čtyři ze čtyř pacientů byli schopni samostatné chůze. Ve skupině 2 bylo 10 pacientů schopno samostatného sedu a dva samostatné chůze [6].
Data z praxe
V České republice je OA dostupný od května 2020. Dva pacienti byli na našem pracovišti léčeni ještě před evropskou registrací v rámci specifického léčebného programu. Aktuálně bylo v ČR léčeno okolo 20 pacientů. V našem centru jsme k srpnu 2021 léčili 12 pacientů ve věku od osmého do 35. měsíce a s hmotností od 7,9 kg do 12,6 kg. Delší než roční interval od podání OA mají prozatím dva pacienti se SMA typu 2, oba mají z léčby prospěch, jsou schopni se sami posadit a nově rovněž schopni samostatného stoje. Ze závažných nežádoucích účinků během prvních tří měsíců po podání jsme pozorovali u dvou pacientů hepatopatii se zvýšením hodnot jaterních transamináz (více než 20 nad normu), u jednoho z těchto pacientů byla hepatopatie provázena poruchou koagulace s nutností léčby.
Jiné alternativy léčby spinální svalové atrofie
Aktuálně jsou kromě OA registrovány další dva kauzální přípravky, nusinersen a risdiplam. Oba přípravky zvyšují tvorbu chybějícího proteinu SMN modulací transkripce RNA SMN2, z tohoto důvodu musejí být podávány opakovaně, celoživotně. Nusinersen je nejstarším registrovaným kauzálním lékem pro SMA na trhu, jeho výhodou je aktuálně desetiletá zkušenost a velký počet (více než 10 000) léčených pacientů. Jelikož lék nepřestupuje hematoencefalickou bariéru, je podáván v pravidelných intervalech intratekálně [15]. Nusinersen má v ČR úhradu pro všechny typy i věkové skupiny pacientů se SMA.
Nově, v srpnu 2020 v USA a v březnu 2021 v EU, byl registrován risdiplam, který má výhodu perorálního podání [16]. Tento lék je indikován pro všechny pacienty se SMA s vazbou na SMN1 starší dvou měsíců, kteří mají čtyři nebo méně kopií SMN2. Ke srovnání efektu nusinersenu, risdiplamu a OA není prozatím dostatek dat, neexistují přímé srovnávací studie. Nedostatek dat a finanční náročnost léčby jsou hlavními důvody, proč zatím není odbornými společnostmi indikována léčba kombinovaná (www.detskaneurologie.cz).
Kauzální léčba zásadním způsobem mění prognózu pacientů se SMA. Nejdůležitějším faktorem predikce efektu je časnost zahájení terapie. Tento faktor je dán počtem ještě zachovalých motoneuronů v době počátku léčby. Je však nutné vědět, že terapie u již symptomatických pacientů tyto nemocné nevyléčí, ale mírně zlepšuje motorické dovednosti a zejména stabilizuje stav a zabraňuje progresi nemoci. Před zahájením léčby symptomatických pacientů by proto měl vždy proběhnout pohovor s nemocným či s jeho rodiči o očekávaném efektu terapie, u nejtěžších klinických forem je ke zvážení také paliativní péče [10]. Nadějí do budoucna je možnost presymptomatické léčby na podkladě novorozeneckého screeningu SMA [17]. Od ledna 2022 je v ČR plánováno spuštění pilotního celorepublikového projektu novorozeneckého screeningu.
Seznam použité literatury
- [1] Mailman MD, Heinz JW, Papp AC, et al. Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2. Genet Med 2002; 4: 20–26.
- [2] Verhaart IEC, Robertson A, Wilson IJ, et al. Prevalence, incidence and carrier frequency of 5q‑linked spinal muscular atrophy – a literature review. Orphanet J Rare Dis 2017; 12: 124.
- [3] Ogino S, Wilson RB, Gold B. New insights on the evolution of the SMN1 and SMN2 region: simulation and meta‑analysis for allele and haplotype frequency calculations. Eur J Human Genetics 2004; 12: 1015–1023.
- [4] New Zolgensma data demonstrate age‑appropriate development when used early, real‑world benefit in older children and durability 5+ years post‑treatment. Mar 15, 2021. Dostupné na: www.novartis.com/news/media‑releases/new‑zolgensma‑data‑demonstrate‑age‑appropriate‑development‑when‑used‑early‑real‑world‑benefit‑older‑children‑and‑durability‑5‑years‑post‑treatment
- [5] Powell SK, Rivera‑Soto R. Viral expression cassette elements to enhance transgene target specificity and expression in gene therapy. Discov Med 2015; 19: 49–57.
- [6] EPAR Zolgensma. Dostupné na: www.ema.europa.eu/en/medicines/human/EPAR/zolgensma
- [7] SPC Zolgensma. Dostupné na: https://www.sukl.cz/modules/medication/detail.php?code=0246243&tab=texts
- [8] FDA Product Information Zolgensma. Dostupné na: www.fda.gov/vaccines‑blood‑biologics/zolgensma
- [9] Zápis z jednání ve věci organizace péče u pacientů se spinální svalovou atrofií (SMA). Praha 15. 7. 2020, Ústředí VZP ČR.
- [10] Kirschner J, Butoianu N, Goemans N, et al. European ad‑hoc consensus statement on gene replacement therapy for spinal muscular atrophy. Eur J Paediatr Neurol 2020; 28: 38–43.
- [11] Glanzman AM, Mazzone E, Main M, et al. The Childrenʼs Hospital of Philadelphia Infant Test of Neuromuscular Disordes (CHOP INTEND): test development and reliability. Neuromuscul Disord 2010; 20: 155–161.
- [12] Day JW, Finkel RS, Chiriboga CA, et al. Onasemnogene abeparvovec gene therapy for symptomatic infantile‑onset spinal muscular atrophy in patients with two copies of SMN2 (STR1VE): an open‑label, single‑arm, multicentre, phase 3 trial. Lancet Neurol 2021; 20: 284–293.
- [13] Farrar MA, Vucic S, Johnston HM, et al. Pathophysiological Insights Derived by Natural History and Motor Function of Spinal Muscular Atrophy. J Pediatr 2013; 162: 15–159.
- [14] Mendell JR, Al‑Zaidy S, Shell R, et al. Single‑Dose Gene‑Replacement Therapy for Spinal Muscular Atrophy. N Engl J Med 2017; 377: 1713–1722.
- [15] Stein CA, Castanotto D. FDA‑Approved Oligonucleotide Therapies in 2017. Mol Ther 2017; 25: 1069–1075.
- [16] Poirier A, Weetall M, Heinig K, et al. Risdiplam distributes and increases SMN protein in both the central nervous system and peripheral organs. Pharmacol Res Perspect 2018; 6: e00447.
- [17] Dangouloff T, Vrščaj E, Servais L, et al. SMA NBS World Study Group. Newborn screening programs for spinal muscular atrophy worldwide: Where we stand and where to go. Neuromuscul Disord 2021; 31: 574−582.