Biosimilární ustekinumab a jeho terapeutická ekvivalence
Ustekinumab (Stelara) byl schválen Evropskou lékovou agenturou (EMA) i americkým Úřadem pro kontrolu potravin a léčiv (FDA) v roce 2009. Biologická terapie představuje velmi účinnou, ale rovněž nákladnou léčebnou modalitu. Se záměrem snížit náklady na léčbu a zlepšit tak její dostupnost pro pacienty jsou vyvíjeny biosimilární přípravky. V důsledku složité struktury biologik se nejedná o strukturně totožné látky, ale o látky podobné. Biosimilární přípravek by měl k referenčnímu přípravku (RP) prokázat tzv. terapeutickou ekvivalenci, tedy doložit srovnatelnou účinnost, aby mohl být schválen ke stejnému použití. Sledovány a porovnávány jsou účinnost, bezpečnost, snášenlivost a imunogenicita přípravku.
Ustekinumab je plně humánní monoklonální protilátka IgG1, která se váže na interleukin (IL) 12 a IL‑23 prostřednictvím jejich společné proteinové podjednotky p40. Dysregulace IL‑12 a IL‑23 je spojována s psoriázou a dalšími zánětlivými onemocněními. Zabráněním vazby p40 na receptorový protein IL‑12R1, který je exprimován na povrchu imunitních buněk, dochází k modulaci cytokinových drah pomocných Th1 a Th17 buněk a k potlačení prozánětlivých imunitních pochodů.
Donedávna nebyl pro ustekinumab dostupný žádný biosimilární přípravek. Na základě klinického hodnocení, jehož klíčové výsledky prezentujeme v tomto sdělení, byl schválen první biosimilární přípravek (Uzpruvo) [1] k RP (Stelara) [2]. Biologicky podobný přípravek pod označením AVT04 byl vyvíjen ve stejných koncentracích jako RP, se shodnými pomocnými látkami, pro subkutánní (s.c.) a intravenózní (i.v.) aplikaci.
Srovnatelné chování v organismu
Farmakokinetikou AVT04 se podrobně zabývala multicentrická randomizovaná, dvojitě zaslepená, tříramenná studie fáze I (NCT04744363) [3]. Její výsledky demonstrovaly farmakokinetickou podobnost, stejně jako srovnatelnou bezpečnost a imunogenicitu mezi kandidátním biosimilárním přípravkem AVT04, RP schváleným v Evropské unii (EU‑RP) a RP licencovaným ve Spojených státech amerických (US‑RP).
U zdravých dospělých jedinců byly v paralelních ramenech porovnávány farmakokinetické profily AVT04, EU‑RP a US‑RP. Primárně hodnocenými parametry byly maximální plazmatická koncentrace (cmax) a plocha pod křivkou plazmatické koncentrace (AUC0‑∞). Farmakokinetická podobnost byla považována za prokázanou, pokud byly všechny 90% intervaly spolehlivosti (CI) pro poměr geometrických průměrů nejmenších čtverců v prespecifikovaném rozmezí 80–125 %. Sledovány byly rovněž další (sekundární) farmakokinetické parametry; bezpečnost a imunogenicita byly hodnoceny až do 92. dne.
Zdraví jedinci ve věku 18–55 let (n = 298) byli randomizováni v poměru 1 : 1 : 1 k aplikaci jednorázové dávky 45 mg AVT04 (n = 98), EU‑RP (n = 99) nebo US‑RP (n = 97) do oblasti břicha, případně stehna. Krevní vzorky byly odebrány před podáním dávky a následně 12 hodin po dávce a ve dnech 9, 15, 29, 57, 78 a 92.
Po podání jednorázové dávky byly zaznamenány srovnatelné profily plazmatických koncentrací v čase. U všech přípravků bylo možné pozorovat fázi absorpce se zvyšováním hodnot až do 168 hodin (9.–10. den), po níž následovala postupná eliminace (graf 1). Sledované farmakokinetické parametry byly všechny v předem specifikovaných hranicích bioekvivalence 80–125 %, což dokládá farmakokinetickou podobnost AVT04, EU‑RP a US‑RP (tab. 1). Systémová expozice ustekinumabu byla dle očekávání závislá na tělesné hmotnosti. Ve všech třech léčebných ramenech ukázala systémová eliminace ustekinumabu pomalou clearance, dlouhý terminální poločas a nízký distribuční objem.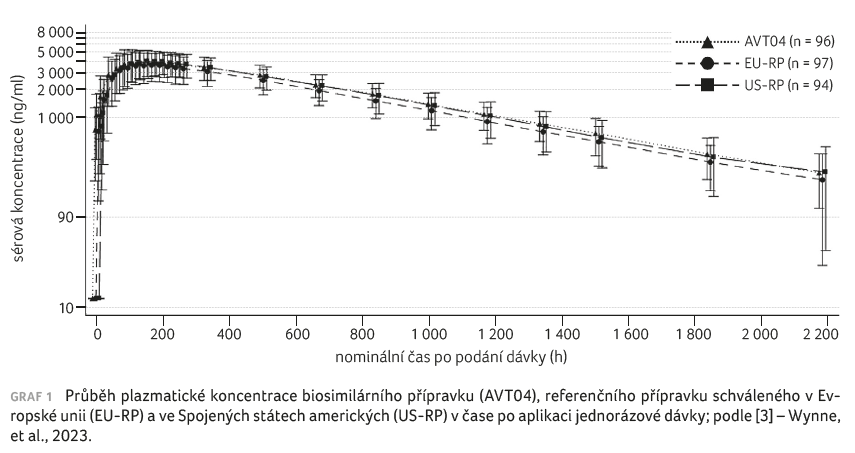
Sledována byla s.c. cesta podání, která představuje hlavní schválenou cestu podání pro RP ustekinumab. Zároveň se tento způsob aplikace považuje za citlivější z hlediska zjišťování rozdílů ve farmakokinetice a imunogenicitě a mohl by poskytnout pohled na potenciální rozdíly.
Imunogenicita byla ve všech třech ramenech srovnatelná, přičemž frekvence detekce protilátek proti léčivu (binding antidrug antibody, ADA) a neutralizujících protilátek (Nab, neutralising antibody) byly nižší u AVT04 než u EU‑RP a US‑RP ve všech časových bodech sledování.
Bezpečnostní profil byl ve všech třech léčebných ramenech rovněž srovnatelný. Sledovány byly výskyt, typ a závažnost nežádoucích účinků. Frekvence nežádoucích účinků souvisejících s léčbou (treatment emergent adverse events, TEAE) byla srovnatelná napříč léčebnými rameny (68,4 % ve skupině AVT04, 67,7 % ve skupině EU‑RP, 71,1 % ve skupině US‑RP) a v souladu s údaji o RP. Nejčastěji hlášenými příhodami byly infekce horních cest dýchacích, bolest hlavy a reakce v místě aplikace.
Terapeutická ekvivalence
Klinické hodnocení fáze III (NCT04930042) publikované Feldmanem a kol. [4] doložilo terapeutickou ekvivalenci AVT04. Studie porovnávala účinnost, bezpečnost, snášenlivost, farmakokinetiku a imunogenicitu mezi AVT04 a EU‑RP u pacientů se středně těžkou až těžkou chronickou ložiskovou psoriázou. Tato multicentrická dvojitě zaslepená 52týdenní studie zahrnovala dvě fáze: primární hodnocení účinnosti (1.–15. týden) a hodnocení dlouhodobé účinnosti a bezpečnosti (16.–52. týden). Pacienti byli randomizováni v poměru 1 : 2 k podávání AVT04 nebo RP. Pacienti s tělesnou hmotností ≤ 100 kg dostávali dávku 45 mg s.c., pacienti s tělesnou hmotností > 100 kg 90 mg s.c. (2 injekce) v den 1 (týden 1), týden 4 a dále každých 12 týdnů (týden 16, 28 a 40). V 16. týdnu pacienti odpovídající na léčbu AVT04 (≥ 50% zlepšení Psoriasis Area and Severity Index [PASI]) pokračovali v aplikaci AVT04, zatímco pacienti, kteří dostávali RP, byli znovu randomizováni v poměru 1 : 1 k podávání AVT04 nebo nadále RP. Pacienti neodpovídající na léčbu do týdne 28 již další léčbu nedostali. Demografické charakteristiky mezi léčebnými skupinami byly vyvážené. Uspořádání studie shrnuje obrázek 1.
 Primárním cílovým ukazatelem bylo procentuální zlepšení PASI od výchozího stavu do týdne 12. Terapeutická ekvivalence byla prokázána, pokud byl interval spolehlivosti pro adjustovaný rozdíl v mezích ekvivalence ±10 % (90% CI).
Primárním cílovým ukazatelem bylo procentuální zlepšení PASI od výchozího stavu do týdne 12. Terapeutická ekvivalence byla prokázána, pokud byl interval spolehlivosti pro adjustovaný rozdíl v mezích ekvivalence ±10 % (90% CI).
Z 581 původně randomizovaných pacientů (194 k AVT04, 387 k RP) jich do týdne 16 setrvalo 575 (99 %), do konce studie 544 (94 %). Ve věku do 65 let bylo 94,3 % pacientů, 62,7 % tvořili muži a 99,3 % zařazených nebylo hispánského ani latino původu.
Studie splnila svůj primární cíl. Procentuální zlepšení PASI pro AVT04 dosáhlo 87,3 %, u RP 86,8 % (CI –2,14 %; 3,01 %). Odpověď PASI se od začátku léčby až do týdne 16 kontinuálně zvyšovala srovnatelně v obou hodnocených skupinách a v dalším období sledování zůstala míra odpovědi setrvalá (graf 2). Při hodnocení dlouhodobé účinnosti (16.–52. týden) bylo zjištěno, že procentuální zlepšení PASI bylo srovnatelné mezi pacienty, kteří přešli z RP na AVT04, a těmi, kteří pokračovali v předchozí léčbě (AVT04/AVT04 a RP/RP).
Analýza podskupin procentuálního zlepšení PASI od výchozího stavu do týdne 12 neodhalila žádné významné rozdíly dle tělesné hmotnosti, předchozí biologické léčby psoriázy, věku, pohlaví, stavu ADA nebo Nab. Homogenita účinku léčby napříč různými podskupinami je jednou ze silných stránek klinického hodnocení, stejně jako vysoká míra odpovědi prokázaná zlepšením PASI o ≥ 50 % u všech pacientů ve studii ve 28. týdnu.
Při pohledu na farmakokinetiku byly údolní (trough) sérové koncentrace ve všech léčebných skupinách srovnatelné po celou dobu sledování (graf 3).
Z hlediska imunogenicity mělo v 16. týdnu 49 (25,4 %) pacientů ve skupině AVT04 a 184 (48,2 %) pacientů ve skupině RP přítomné ADA. Z nich 13 (26,5 %) pacientů ve skupině AVT04 a 57 (31,0 %) pacientů ve skupině RP mělo NAb. Na konci studie byly ADA prokázány u 39 (21,2 %) pacientů ve skupině AVT04/AVT04, u 56 (31,5 %) pacientů ve skupině RP/AVT04 a u 48 (26,7 %) pacientů ve skupině RP/RP. Z toho 13 (33,3 %) pacientů ve skupině AVT04/AVT04, 10 (17,9 %) pacientů ve skupině RP/AVT04 a 11 (22,9 %) pacientů ve skupině RP/RP mělo NAb. Numerické rozdíly ve frekvenci detekce ADA a NAb neměly žádný klinicky významný dopad na účinnost studijní léčby, její bezpečnost nebo farmakokinetický profil., a blokuje tak jejich vazebné interakce s ligandy PD‑L1 a PD‑L2.")
Zjištěný bezpečnostní profil potvrdil předchozí klinická data. Do 16. týdne hlásilo 10 (5,2 %) pacientů celkem 13 TEAE souvisejících s léčbou ve skupině AVT04 a 37 (9,6 %) pacientů uvedlo 39 TEAE souvisejících s léčbou ve skupině RP. Do konce studie zůstaly bezpečnostní parametry z velké části nezměněny a srovnatelné mezi léčebnými rameny i po změně léčby v týdnu 16. Hlášené TEAE byly od týdne 16 do konce studie numericky vyšší ve skupinách RP/AVT04 a RP/RP oproti skupině AVT04/AVT04. Většina TEAE byla mírné až střední závažnosti bez ohledu na zařazení do léčebné skupiny.
Účinnost, bezpečnost a farmakokinetické profily byly srovnatelné napříč léčebnými rameny po celou dobu trvání studie (i po změně léčby). Očekává se, že biosimilars zajistí podobnou klinickou odpověď na léčbu jako RP s možností přechodu (switch) mezi nimi bez dodatečných rizik. Vzhledem k tomu, že AVT04 je vyvíjen jako biologicky podobný přípravek, lze u AVT04 očekávat nežádoucí účinky podobné těm, které byly hlášeny pro RP. Oproti RP nebyly zaznamenány žádné nové bezpečnostní signály.
Při vývoji přípravku je třeba vzít v úvahu možnou imunogenicitu, která může vést k posunu nebo ztrátě biologické účinnosti a bezpečnosti terapie. Již dříve bylo v klinických studiích prokázáno, že ustekinumab (RP) má nízkou incidenci tvorby protilátek, která se po opětovném vystavení léčbě nezvýšila. Navíc se nezdálo, že by přítomnost protilátek proti ustekinumabu ovlivnila jeho terapeutický účinek [5]. Z tohoto pohledu neprokázala tato klinická hodnocení žádné rozdíly biosimilárního přípravku oproti RP a výskyt protilátek proti ustekinumabu neměl žádný klinicky významný dopad [4].
Závěr
Prezentovaná klinická hodnocení prokázala terapeutickou ekvivalenci mezi AVT04 a RP u pacientů se středně těžkou až těžkou chronickou psoriázou s podobnou bezpečností a snášenlivostí. Tato evidence také podporuje vědecké zdůvodnění potenciální extrapolace na další schválené indikace ustekinumabu.
Literatura
[1] Souhrn údajů o přípravku Uzpruvo. Dostupné na: https://www.sukl.cz
[2] Souhrn údajů o přípravku Stelara. Dostupné na: https://www.sukl.cz
[3] Wynne C, Hamilton P, McLendon K, et al. A randomized, double‑blind, 3‑arm, parallel study assessing the pharmacokinetics, safety, tolerability and immunogenicity of AVT04, an ustekinumab candidate biosimilar, in healthy adults. Expert Opin Investig Drugs 2023; 32: 417–427.
[4] Feldman SR, Reznichenko N, Berti F, et al. Randomized, double‑blind, multicenter study to evaluate efficacy, safety, tolerability, and immunogenicity between AVT04 and the reference product ustekinumab in patients with moderate‑to‑severe chronic plaque psoriasis. Expert Opin Biol Ther 2023; 23: 759–771.
[5] Assessment Report for Stelara. Dostupné na: https://www.ema.europa.eu/en/documents/assessment‑report/stelara‑epar‑public‑assessment‑report_en.pdf