Farmakogenetika, účinek léku a lékové interakce
Podání léku nemocnému je vždy do jisté míry experimentem. Na výsledném terapeutickém efektu se uplatní zejména individuální vnímavost, která závisí na řadě faktorů, zejména na genotypu nemocného. Rozhodující jsou polymorfismy uplatňující se v etiopatogenezi choroby, polymorfismy v oblasti metabolismu léku i polymorfismy cílových receptorů zodpovídajících za vlastní účinek léku. Tak například při podání B-blokátoru metoprololu se na výsledném antihypertenzním účinku uplatní typ hypertenze, resp. aktivita jednotlivých regulačních mechanismů (sympato-adrenálního, renin-angiotenzinového či aktivita renálních tubulárních systémů), aktivita systému CYP 2D6, resp. jeho polymorfismus ovlivňující metabolismus lipofilních B-blokátorů, i periferní aktivita pre- a postsynaptických adrenergních receptorů, která je opět ovlivněna jejich polymorfismem.
Druhou důležitou okolností rozhodující o účinku léku u daného nemocného je komedikace. Všichni víme, že řada léků se vzájemně ovlivňuje. Tyto lékové interakce mohou vznikat na úrovni farmakokinetických i farmakodynamických vlastností. O významu interakcí svědčí údaje získané z analýzy národních registrů. Tak například rozborem registru v USA bylo zjištěno, že nežádoucí účinky léků jsou na 4. až 6. místě mezi nejčastějšími příčinami úmrtí [1]. Předpokládá se přitom, že lékové interakce jsou příčinou více než poloviny závažných nežádoucích příhod [2]. Z jiné analýzy vyplývá, že rozhodující podíl na vzniku závažných lékových interakcí mají polymorfismy v metabolismu relativně nevelkého počtu „problematických" léků či lékových skupin [3]. Bohužel povědomí lékařů o takovýchto „rizikových" či přímo „kontraindikovaných" komedikacích je nízké. Jak vyplývá z analýzy více než tří milionů receptů Zdravotní pojišťovny Škoda provedené v roce 2004 dr. Suchopárem, nejméně 15 % vzájemných kombinací předepsaných léků bylo minimálně „velmi rizikových" [4]. Rozhodující většina, tj. více než 90 % těchto nevhodných kombinací, se týkala kardiovaskulárních léků. Také proto je věnován tento přehled kardiologům.
Mluvíme-li o metabolismu léků a o farmakogenetice, ujasněme si nejprve základní pojmy. Genetická informace je uchována v kódující DNA, polymeru obsahujícím páry nukleotidů. Trojice těchto nukleotidů určuje pořadí každé jedné z 20 aminokyselin v řetězci proteinu. Soubor těchto jednotlivých informací nazýváme genomem. Lidský genom představuje ohromné množství více než 3 gigabází v DNA (sekvencí párů nukleotidů). Menší část genomu je tvořena kódujícími úseky (tzv. introny), které určují asi 32 tisíc genů. Předpokládá se však, že necelá polovina kóduje vlastní proteinový řetězec. Díky různým posttranskripčním úpravám však tento relativně malý počet genů výsledně determinuje více než 100 tisíc bílkovinných molekul. I když je DNA neobvykle stabilní strukturou, mohou při udržování a předávání informace pomocí sekvence párů bází DNA vznikat chyby. Naprostá většina těchto chyb, lépe řečeno odchylek, je dána odlišností jednoho páru, tedy bodovou mutací. Podstatné je, jak se tato odchylka v pořadí odrazí na funkci proteinu. Změna v genotypu může postihnout také oblasti DNA, které nekódují vlastní proteiny (tzv. intron). Tyto oblasti pravděpodobně řídí přepis DNA a syntézu vlastních proteinů. Zdá se, že změny kódu v intronu mohou vést ke zvýšenému či sníženému přepisu genu (expresi), a snad i k multiplikaci jednotlivých genů, k tzv. multiplikaci alel.
Jaký je dopad těchto bodových mutací? Závažné poruchy syntézy či exprese důležitých proteinů (event. lipoproteinů či glykoproteinů) bývají letální. Méně závažné, nicméně pro svého nositele nevýhodné mutace jsou eliminovány vývojem. Nezávažné odchylky funkce, které se neprojeví znevýhodněním v reprodukci, však mohou být v populaci časté. V našich více než 30 tisících genů jsou známy nejméně 2 miliony takovýchto odchylek neboli polymorfismů.
Obecně je polymorfismus definován jako odchylka v sekvenci DNA, která je přítomna nejméně v 1% populace, často však bývá rozšířena u daleko většího počtu jedinců, tj. v desítkách procent. Polymorfismus nemusí vést k odchylce ve funkci, v řadě případů však vede ke snížené aktivitě proteinu, například enzymů, receptorů nebo transportních proteinů, vzácněji naopak může být aktivita větší. Významem těchto polymorfismů ve farmakologii se zabývá jak farmakogenetika, tak farmakogenomika. Farmakogenetika studuje dopad těchto polymorfismů v enzymech aktivujících či degradujících léky, v receptorech určujících odpověď na lék či v polymorfismu enzymů určujících syntézu substrátu pro daný lék. Daleko užší pojem – farmako-genomika – se pak zabývá individuální schopností jedince zacházet s léčivem, tj. variacemi v absorpci, aktivaci, distribuci, vylučování či interakci s cílovou strukturou (např. s receptorem).
Polymorfismus se uplatní na mnoha úrovních: v samotné etiopatogenezi chorob s monogenní či polygenní etiologií, v odpovědi na léčbu, v odpovědi na podaný lék, v resorpci a metabolismu daného léku i v riziku vzájemné interakce mezi léky.
Jedním z hlavních úkolů farmakogenetiky je genotypizace pochodů ovlivňujících kinetiku léků. Zjištění genotypu, a tím i určení aktivity systémů účastnících se na procesu vstřebávání léku, na jeho aktivaci či degradaci při průchodu játry či systémovým řečištěm, na jeho distribuci v organismu, na jeho průchodu hematoencefalickou či placentární bariérou či na jeho degradaci a vyloučení z organismu má bezprostřední praktický význam. Organismus má k dispozici v zásadě tři řady systémů, které zajišťují osud léku v organismu. Jejich zapojení má zpravidla jediný cíl: usnadnit eliminaci z organismu, tj. zejména zvýšit jeho rozpustnost ve vodě, aby mohl být ledvinami vyloučen. Z praktických důvodů dělíme tyto systémy podle typu reakce, které zajišťují. Enzymy I. fáze mění charakter molekuly hydroxylací, enzymy II. fáze navazují vnesením hydrofilní skupiny (nejčastěji glukuronové či acetylové). Konečně ve III. fázi je lék vyloučen specifickými transportními systémy z organismu do moče či žluče.
a) Cytochromový systém P-450 (CYP)
Tento typ reakce změní molekulu oxidací, redukcí či hydrolýzou, aby molekula byla snáze konjugována s endogenními látkami usnadňujícími rozpustnost potřebnou ke vstřebání, či naopak k vyloučení. Reakce I. fáze jsou zprostředkovány pozoruhodnou rodinou enzymů cytochromového systému P-450 (CYP).
CYP a lékové interakce
Společnou cestou biodegradace nebo bioaktivace podstatné části běžně užívaných léků je změna funkčních skupin výchozí molekuly cytochromovým systémem. Cílem je zvýšit hydrofilii mateřské molekuly, a tím snížit resorpci ve střevě nebo zvýšit exkreci látky do moče či do žluče. Cytochromový systém P-450 je rodina enzymů, oxygenáz, obsahujících protoporfyrinové jádro (hemoproteiny), které přidáním či změnou funkční skupiny mění polaritu endogenních i exogenních molekul. Tak se mění rozpustnost substance, dochází k degradaci, případně k aktivaci. Vlastní změna funkční skupiny je variabilní a závisí na vlastnostech molekuly substrátu i typu cytochromu. Charakteristické je výrazné překrývání účinku na cílový substrát (jedna molekula může být modifikována několika enzymy systému) i široké spektrum substrátů pro každý z téměř 500 jednotlivých izoenzymů. Izoenzymy jsou lokalizovány zejména v játrech, ve střevě, v plicích a v ledvinách, kde jsou vázány na membrány endoplazmatického retikula.
Jak již bylo řečeno, úlohou systému je biodegradace endogenních substancí (např. steroidů, prostanoidů či regulačních proteinů) i exogenních substancí (např. flavonoidů, alkaloidů či léků). Změna molekuly substrátu může vést též ke konverzi na aktivní substanci, tj. k aktivaci (příkladem je konverze inaktivních forem vitaminů na aktivní či cholesterolu na steroidní hormony). Oxygenázy CYP též usnadňují resorpci látky ze zažívacího traktu. Systém se rozhodující měrou podílí se na „first-pass efektu", tj. modifikaci molekuly při prvním průchodu játry. Vztah enzymu a cílové molekuly je oboustranný. Také cílová molekula může ovlivnit aktivitu cytochromového systému. Podle vzájemné interakce rozlišujeme tři kategorie. První kategorií je substrát, ten neovlivňuje aktivitu systému CYP. Druhou je induktor, ten naopak zvyšuje aktivitu systému CYP. Konečně třetím případem je, že inhibitor blokuje aktivitu systému CYP. Tato inhibice může být reverzibilní (kompetice) či ireverzibilní (trvalá blokáda enzymu). Jednotlivé izoenzymy cytochromového systému jsou aktivovány či inhibovány odlišnými molekulami. Vzájemné interakce substrátu a induktoru či inhibitoru ve střevě, v játrech či v plicích zásadním způsobem mění presystémovou a systémovou dostupnost léku, tj. množství léku, které se dostane do místa svého působení.
Polymorfismus systému CYP
Na úrovni cytochromového systému je dnes odhaleno téměř 80 různých polymorfismů. Některé vedou ke snížení aktivity enzymu až k jeho naprosté neúčinnosti, jiné naopak efekt enzymu zvyšují. Závisí jednak na charakteru substituce, jednak na tom, zda má jedinec daným polymorfismen postiženy oba geny neboli obě alely. Homozygotní postižení bývá závažnější. Je logické, že takováto změna se musí odrazit v osudu léku v organismu, tj. ve farmakokinetice. Podle genotypu pro daný CYP (např. 2D6 či 2C9) pak rozlišujeme populaci na pomalé, střední, rychlé a ultrarychlé metabolizátory. Běžná dávkovací schémata léků pak často vyhovují pro střední, event. rychlé metabolizátory, u pomalých dochází ke kumulaci až k toxickým hladinám, či naopak u velmi rychlých metabolizátorů hladina léku po většinu dávkovacího období nedosáhne úrovně terapeutické hladiny a léčba je neúčinná. Rychlost degradace některých léků daným systémem se pak podle genotypu může odlišovat až tisícinásobně. Naštěstí však díky překrývání substrátů bývá lék odbourán též jiným systémem a v praxi se tak interindividuální koncentrace léku odlišují o jeden, maximálně o dva řády [3].
Za metabolismus rozhodující většiny kardiovaskulárních léků jsou zodpovědné tři izoenzymy CYP: 3A4, 2C9 a 2D6. Poslední dva, tj. CYP 2C9 a 2D6, mají velmi výrazný polymorfismus. Pro klinika je důležité, že některé typy polymorfismu se vyskytují v populaci až v desítkách procent, a musíme tedy jejich přítomnost zohlednit v léčebné praxi. Problémem však je, že stanovení polymorfismu je sice možné, nicméně pro širší užití kapacita zatím nestačí. Proto nám zbývá pouze odhadovat výslednou hladinu léku z farmakologického účinku: např. stanovením hladiny INR při léčbě warfarinem či sledováním srdeční frekvence při léčbě b-blokátory. V současné době je patrný rozmach stanovování základních polymorfismů CYP ve všech vyspělých zemích, sami tuto možnost máme již druhým rokem.
CYP 3A4: Nejdůležitějším enzymem systému je CYP 3A4, touto cestou je metabolizováno asi 55 % všech užívaných léků (tab. 1). Lokalizován je především v játrech, tenkém střevě, plicní cirkulaci či v ledvinách. Klinicky nejvýznamnějšími substráty jsou některé statiny (atorvastatin, simvastatin a lovastatin), blokátory kalciového kanálu, makrolidová antibiotika, steroidy či některá antiarytmika (např. amiodaron) nebo některá cytostatika. Naštěstí u tohoto exponovaného CYP nebyly odhaleny funkčně významné polymorfismy. O to významnější jsou však interakce na tomto systému. Řada běžně užívaných léků či potravy totiž zásadním způsobem inhibuje nebo naopak stimuluje aktivitu enzymů této skupiny (tab. 1).
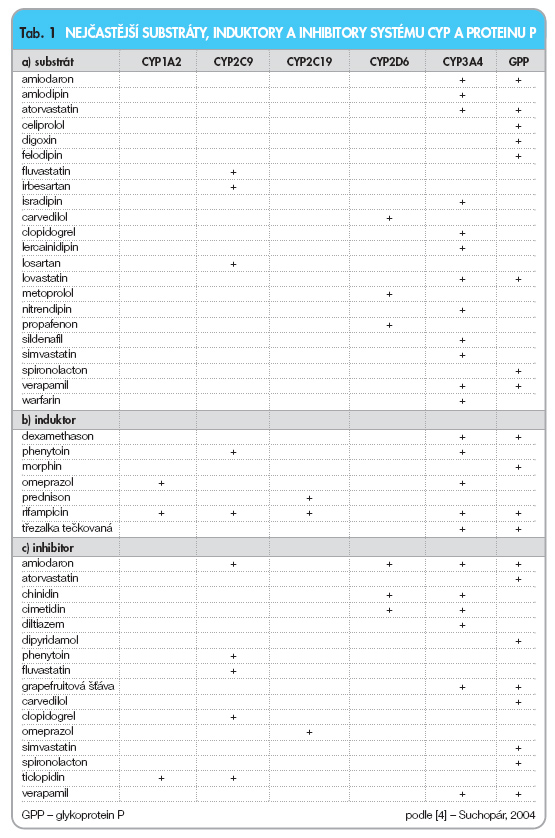
K nejdůležitějším inhibitorům CYP 3A4 patří antiulcerózum cimetidin, řada makrolidových antibiotik (např. erythromycin, clarithromycin), metronidazol, methylprednisolon, antimykotika, některé blokátory kalciového kanálu (diltiazem a verapamil) či grapefruitová šťáva a mírnými inhibitory jsou i některé flavonoidy z vína. Induktory zvyšujícími aktivitu jsou například dexamethason, phenobarbital, omeprazol, antidiabetika glitazonové řady (pioglitazon a troglitazon) či třezalka.
Ke klinicky nejvýznamnějším interakcím patří běžně podávaná kombinace inhibitorů CYP 3A4 diltiazemu či verapamilu se substráty, jakými jsou statiny. Tato kombinace je na jedné straně častá, v analýze preskripce v posledních letech byla např. kombinace simvastatinu s verapamilem či s diltiazemem předepsána asi ve 2,4 % všech předpisů, na straně druhé je klinicky závažná [4]. Hladiny statinů mohou při těchto kombinacích stoupat i více než o řád [5]. Riziko myopatií pak výrazně stoupá. Podobně ke zvýšení hladin statinů či jiných substrátů CYP 3A4 vede kombinace s ostatními výše uvedenými inhibitory (graf 1). Naopak pije-li pacient pravidelně např. „neškodný" čaj z třezalky, můžeme očekávat snížení terapeutického efektu indukcí enzymu a rychlého odbourání léku.
O tom, že inhibice CYP 3A4 je významná nejen pro metabolismus, resp. degradaci v játrech, ale též pro resorpci léku, nás přesvědčí druhý příklad. Podívejme se na blokátory kalciového kanálu s nízkou biologickou dostupností, např. felodipin s dostupností kolem 15 %. Ve střevní sliznici i v játrech je léčivo degradováno CYP 3A4 a do systémové cirkulace se dostane jen 15 % perorálně podaného léku. Inhibujeme-li cytochromový systém například komedikací cimetidinem nebo vypije-li pacient sklenici grapefruitové šťávy, pak nedojde k presystémové degradaci a dostupnost stoupne až na 90 %, tj. stejně jako bychom podali šestinásobnou dávku [6]. Výsledná hypotenze je nasnadě. Pro praxi z tohoto příkladu vyplývá závěr: Chceme-li se vyvarovat tohoto typu interakcí, indikujme našim nemocným přednostně léky s vyšší biologickou dostupností. V případě blokátorů kalciového kanálu má například amlodipin dostupnost kolem 60 %, zde její zvýšení na 90 % bude klinicky nevýznamné.
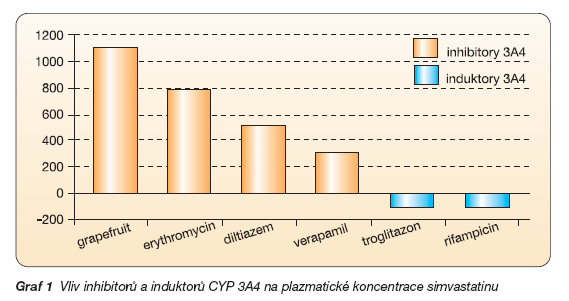
CYP 2D6: Největší počet klinicky významných polymorfismů byl odhalen v podskupině CYP 2D6. Tento izoenzym metabolizuje asi 25–30 % léků, například lipofilní b-blokátory (metoprolol a carvedilol), některá antiarytmika (např. propafenon či mexiletin), většinu antidepresiv či antipsychotik (tab. 1). Podle aktivity výsledného genotypu CYP 2D6 je možno odlišit pomalé metabolizátory s dvěma nefunkčními alelami, střední s jednou nefunkční alelou, rychlé (s dvěma funkčními alelami) a ultrarychlé metabolizátory (s více kopiemi genu). Takováto multiplikace alel může dosáhnout i více než desetinásobného zdvojení, rychlost degradace je pak enormní [7]. U pomalých metabolizátorů, tvořících 6–10 % naší populace, dochází ke kumulaci, k prodlouženému účinku léku a k častějším nežádoucím účinkům. Popsány jsou např. toxické hladiny propafenonu či mexiletinu s arytmiemi či se zvracením. Naopak u velmi rychlých metabolizátorů, kterých je v naší populaci asi 5 %, běžnými dávkami nedosáhneme léčebného účinku.
V praxi je důležitá velmi variabilní hladina lipofilních b-blokátorů. V populaci se setkáváme s až desetinásobnými rozdíly koncentrace metoprololu či carvedilolu mezi osobami s různými genotypy. Podobně je hladina lipofilních b-blokátorů citlivá na komedikaci inhibitory a induktory CYP 2D6 (graf 2).
Naštěstí u b-blokátorů si lehce s léčebnou dávkou poradíme, stačí se orientovat podle srdeční frekvence. Optimální dávkou bychom měli dosáhnout frekvence kolem 50–60 tepů/minutu. Druhou možností je volit b-blokátor hydrofilní (např. bisoprolol či atenolol). Mluvíme-li o lipofilních b-blokátorech, je možno zmínit ještě zajímavou interakci s některými antiarytmiky. Tak amiodaron či propafenon, které jsou často s b-blokátory komedikovány, inhibují CYP 2D6, a zvyšují tak hladinu b-blokátoru. Toto zvýšení se projeví zejména u rychlých metabolizátorů, naopak u pomalých se projevit nemůže pro již tak nízkou aktivitu systému. Tato interakce by se mohla podílet na výrazném bradykardizujícím účinku, který můžeme někdy pozorovat po nasazení amiodaronu k již vytitrované dávce b-lytika.
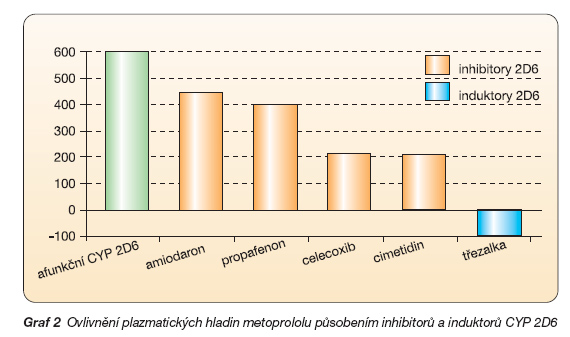
Daleko obtížněji se však budeme orientovat ve výsledném účinku u antidepresiv. Nepřiměřený útlum na jedné straně, či naopak selhání účinku na straně druhé budou projevy, které můžeme očekávat u obou krajních genotypů. Jak je vidět z grafu 3, je například u antiarytmika propafenonu dávkování založené na populačních studiích nevyhovující pro pomalé, část intermediálních a všechny ultrarychlé metabolizátory.
Prakticky to znamená, že 1 až 1,5 milionu osob v Česku můžeme považovat z hlediska vhodného dávkování léků transformovaných CYP 2D6 za jaksi „zapomenutou" populaci.
U pomalých metabolizátorů CYP 2D6 se můžeme setkat také s opačným fenoménem. Tento enzym je nutný k transformaci proléčiva codeinu na analgeticky působící morphin. Podobná situace je u tramadolu, i zde je nutná biotransformace k zajištění analgetického efektu. U pomalých metabolizátorů účinek těchto léků bohužel selhává, nevytvoří se dostatečná hladina aktivního metabolitu.
CYP 2C9: Tento třetí nejvýznamnější komplex je zodpovědný za biotransformaci asi 10 % léků. Zajišťuje například degradaci warfarinu, řady nesteroidních antirevmatik (např. koxibů, ibuprofenu, indometacinu), perorálních antidiabetik (typu derivátů sulfonylurey) či za transformaci antagonistů receptorů AT1 – sartanů (losartanu, irbesartanu či valsartanu) (tab. 1). I u CYP 2C9 jsou identifikovány tři alely vedoucí ke snížené aktivitě systému. Genotypy nesoucí například alelu CYP 2C9*2 a CYP 2C9*3 mají zásadní význam, neboť vedou k pěti-, resp. 27násobnému snížení clearance substrátů, zejména warfarinu či perorálních antidiabetik, ve srovnání s běžnou alelou. Kumulace warfarinu je u těchto osob spojena s častými krvácivými komplikacemi a účinné dávky jsou velmi nízké. Přiměřené dávkování warfarinu se pak u jednotlivých typů metabolizátorů liší i o řád. Podobně prodloužení účinku jiného substrátu, například tolbutamidu, je spojeno s rizikem hypoglykemických stavů. Frekvence výskytu těchto genotypů je přitom v naší populaci častá, typ CYP 2C9*2 se objevuje asi u 10–20 % jedinců a typ CYP 2C9*3 u 6–9 %.
b) Membránové transportní systémy
Transportní membránové proteiny umožňují přechod přes buněčné membrány. Řada léků (či obecněji xenobiotik) má totiž část molekuly hydrofilní, čímž je zajištěna rozpustnost ve vodě, a část molekuly natolik lipofilní, že je umožněna pasivní difuze skrz lipidovou dvojvrstvu buněčné membrány. U podobných molekul je nebezpečí, že budou ve vysoké míře vstřebány a transportovány do organismu, jemuž pak hrozí nebezpečí poškození. Navíc některé tkáně – mozek, plod či spermie – jsou vůči působení xenobiotik zvláště citlivé. Během vývoje se objevuje mechanismus, který je schopen vyloučit podobné látky ven z organismu, a navíc vytvořit bariéry, které chrání určité tkáně. Příkladem takové bariéry je hematoencefalická bariéra, kdy v endoteliích cév mozku a v neurogliích je systém pump silně aktivován a cizorodé látky, které překročí endoteliální výstelku, jsou aktivně vraceny zpět do krve. Celý systém má uniformní strukturu. Jedná se o jeden řetězec proteinu, který má část procházející spirálovitě buněčnou membránou a tvořící jakýsi pór a část volného řetězce s vazebným místem pro ligand a pro ATP [8]. Právě závislost funkce na energii dodané makroergním fosfátem dala celé skupině jméno: rodina transportních pump ABC (ATP-binding cassette).
V obecnější rovině můžeme do této kategorie transportních mechanismů zařadit i iontové pumpy, které za využití ATP udržují patřičný gradient. Podobné pumpy regulují i transmembranózní přesun řady důležitých organických látek, například glycidů, aminokyselin, cholesterolu, žlučových kyselin či vitaminů. Takovéto pumpy nalezneme v ledvinách, plicích, trávicím traktu, játrech a jinde. Jejich poruchy, nejčastěji se jedná o mutaci proteinu, vedou k řadě chorob, jako je hypertenze, cystická fibróza, některé dyslipidémie či např. Dubinův-Johnsonův syndrom [10].
Dosud bylo odhaleno asi 50 druhů transportních kanálů ABC. Podívejme se na jejich nejdůležitějšího představitele, glykoprotein P. Tento aktivní transportní kanál byl objeven při objasňování příčiny vzniku rezistence nádorových buněk na řadu cytostatik. Byl objeven gen (tzv. MDR – multidrug resistance), jehož aktivace vedla k této rezistenci. Expresí tohoto genu dojde k syntéze glykoproteinu P, který po zabudování do buněčné membrány vede k vylučování cizorodých látek z lipidové membrány buňky ven do extracelulárního prostoru. Právě aktivace tohoto proteinu P je jednou z hlavních příčin vymanění se nádorových buněk z efektu cytostatik po opakované kúře či je podkladem primární rezistence mozkových nádorů k chemoterapii [10]. Glykoprotein P má mnoho vlastností společných s CYP 3A4. Je lokalizován ve stejných tkáních, tj. v tenkém střevě a v játrech, má stejnou úlohu, tj. prevenci vstupu xenobiotik do těla, a v neposlední řadě působí na řadu stejných substrátů.
Podobně jako systém cytochromových oxidáz má i glykoprotein P své substráty, induktory a inhibitory (tab. 1). Substráty bývají zpravidla molekuly s částí lipofilní a s částí hydrofilní, příklady jsou léky se steroidním jádrem (aldosteron, kortisol, prednison, digoxin aj.) nebo například amiodaron, atorvastatin, ciclosporin, cimetidin, diltiazem, erythromycin, morphin, tetracyklin, verapamil. Nutno zdůraznit, že řada substrátů (např. diltiazem či verapamil) je též metabolizována jinými systémy, zejména CYP 3A4. Podobně některé induktory proteinu P (např. phenobarbital, dexamethason či extrakt z třezalky) indukují CYP 3A4 a stejně některé inhibitory (např. diltiazem, verapamil, antimykotika či grapefruitová šťáva) inhibují též CYP 3A4 nebo (chinidin) CYP 2D9.
Také u genu MDR je známo několik mutací vedoucích k polymorfismu glykoproteinu P. Některé vedou ke snížení substrátové specificity či k oslabení až vymizení aktivity glykoproteinu. Významným polymorfismem je C3435T, kdy v evropské populaci je asi čtvrtina osob homozygotními nositeli obou aktivních alel CC, polovina heterozygotů s alelami CT a čtvrtina homozygotů s oběma neaktivními alelami TT.
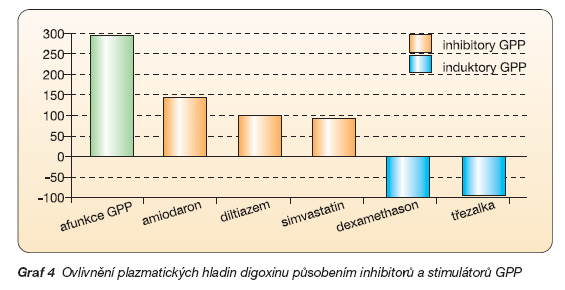
Dopad přítomnosti obou neaktivních alel TT, která je spojena s dysfunkcí glykoproteinu P, je možno demonstrovat například na digoxinu. Ten je substrátem pro glykoprotein P. Nositelé genotypu TT mají zvýšené vstřebávání digoxinu a nižší clearance. Proto při běžném dávkování mohou dosahovat plazmatické hodnoty toxického pásma se všemi průvodními jevy, jakými jsou výskyt arytmií a riziko náhlé smrti. Opakovaně bylo poukázáno, že nositelé této alely by tak měli dostávat řádově poloviční dávku digoxinu. Zevšeobecníme-li, pak by měli dostávat nižší dávky i ostatních léků, které jsou substrátem glykoproteinu P.
Analogickou situaci, jakou je dysfunkce následkem polymorfismu, můžeme navodit podáním inhibitoru glykoproteinu P. V sedmdesátých letech byla popsána závažná interakce při výrazném zvýšení koncentrace digoxinu po podání chinidinu. Vysvětlení se našlo až po objasnění inhibice glykoproteinu P chinidinem, a tím zvýšené biologické dostupnosti digoxinu. V současné době však tolik nehrozí interakce digoxinu s chinidinem, tato komedikace je výjimečná. Daleko závažnější je časté podávání inhibitorů glykoproteinu P amiodaronu, atorvastatinu, lovastatinu, simvastatinu, spironolactonu či carvedilolu spolu s digoxinem u nemocných se srdečním selháním. Jinou častou komedikací u nemocných s fibrilací síní je preskripce inhibitoru glykoproteinu P verapamilu či diltiazemu s digoxinem. Kombinace inhibitoru glykoproteinu P s digoxinem není jistě výhodná, nicméně občas se jí nevyhneme. V takovém případě dávku digoxinu redukujeme a kontrolujeme, zda stále udržujeme terapeutickou hladinu.
Jistě žádný z nás si nepamatuje metabolismus všech léků, nicméně při preskripci některých skupin léků bychom měli „zavětřit" a zvážit, zda nehrozí interakce. Takovými rizikovými skupinami z hlediska inhibice glykoproteinu P jsou například některé statiny, steroidy, antimykotika, makrolidová antibiotika, verapamil, diltiazem, amiodaron či grapefruitová šťáva (graf 4). Na druhé straně je popsáno významné snížení dostupnosti řady léků, substrátů systému, při komedikaci extraktem třezalky, důležitého induktoru systému. Třezalka je přitom běžnou součástí medicinálních čajů, které starší lidé pijí.
c) Cílový receptor působení léku
Každý lék působí zpravidla na jeden či více cílových receptorů. Tyto receptory mohou stimulovat či naopak inhibovat určitý pochod. V současné době známe stovky polymorfismů nejrůznějších receptorů. Snížení či naopak zvýšení odpovědi na podnět díky dysfunkci receptoru může významně ovlivnit odpověď na lék. V některých případech stačí polymorfismus jednoho receptoru k patologické reakci, jindy je nutná souhra polymorfismů dvou či více receptorů.
V kardiologii máme hezký příklad, kdy k patologické odpovědi je nutná souhra mutací na dvou spolupracujících receptorech. Sympatická aktivace myokardu je pod kontrolou presynaptického receptoru ARa2c, který inhibuje uvolnění noradrenalinu z nervového zakončení a z postsynaptického ARb1 zprostředkovávajícího odpověď kardiomyocytu na uvolnění noradre- nalinu. Je znám polymorfismus presynaptického receptoru spojený s jeho nižší tlumivou aktivitou a polymorfismus postsynaptického receptoru spojený naopak s hyperaktivitou. Izolované polymorfismy ARa2c (Del32-325) a ARb1 (arg389), které jsou v populaci asi v 10–20 %, nemění odpověď na stimulaci. Hyperaktivita jednoho je kontrolována útlumem druhého a naopak. Teprve souhra obou polymorfismů dohromady vede k výrazné adrenergní hyperstimulaci myokardu a k mnohonásobnému zvýšení rizika srdečního selhání. Právě u těchto osob je nejúčinnější léčba b-blokátory [11].
V praxi je zdaleka nejčastější kombinace různých polymorfismů v etiologii tzv. multifaktoriálních chorob, jako je hypertenze či aterogeneze.
Jak je patrné z předložených dat, význam farmakogenetiky je široký, projeví se na vstřebávání, distribuci, eliminaci či aktivaci léku, ovlivní aktivaci receptoru nebo stimulaci či inhibici metabolického řetězce. Podobně jako genetická výbava může na osud léku v organismu působit interakce mezi léky. Tyto skutečnosti je potřeba si uvědomit zejména dnes, kdy velká řada nemocných užívá více než 4 léky a kdy riziko lékových interakcí je vysoké. Nyní nastala doba, kdy jednotlivé polymorfismy metabolických a transportních systémů můžeme stanovovat a kdy máme po ruce Kompendium lékových interakcí [4]. Bude jen na nás, abychom tyto informace využili ve prospěch našich nemocných.
Seznam použité literatury
- [1] Lazaru J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalised patients – a meta-analysis of prospective studies. JAMA 1998; 279: 1200–1205.
- [2] Philips KA, et al. Potential role of pharmacogenomics in reducing adverse drug reactions: a systematic review. JAMA 2001; 286: 2270–2279.
- [3] McNamara DM, et al. Pharmacogenomic interactions. Circulation 2001; 103: 1644.
- [4] Suchopár J, et al. Kompendium lékových interakcí Infopharm. Infopharm a.s., Praha, 2004.
- [5] Muck W. Metabolic interactions between mibefradil and HMG-CoA reductase inhibitors: linking in vitro with in vivo information. Br J Clin Pharmacol. 2000; 49 (1): 87–90.
- [6] Takanaga H, Ohmishui A, Matuso H, et al. Pharmacokinetic analysis of felodipine-grapefruit juice interaction based on an irreversible enzyme inhibition model. Br J Clin Pharmacol. 2000; 49 (1): 49–58.
- [7] Ingelman-Sundberg M. Pharmacogenetics of cytochrome P-450 and its applications in drug therapy. Trends Pharmacol Sci 2004; 25 (4): 194–200.
- [8] Borst P, Evers R, Kool M, et al. A family of drug transporters: the Multidrug Resistence-Associated Proteins. J Natl Cancer Inst 2000; 92: 1295–1302.
- [9] Siest G, Ferrari L, Accaoui MJ, et al. Pharmacogenomics of drug affecting the cardiovascular systém. Clin Chem Lab Med 2003; 41 (4): 590–599.
- [10] Sparreboom A, Danesi R, Ando Y, et al. Pharmacogenomics of ABC transporters and its role in cancer chemotherapy. Drug Resistance Updates 2003; 6: 71–84.
- [11] Small KM, Wagoner LF, Levin AM, et al. Synergistic polymorphisms of 1- and 2C-adrenergic receptors and the risk of congestive heart failure. N Engl J Med. 2002; 347 (15):1135–1142.
- [12] McNamara DM. Pharmacogenomic interactions. Circulation 2001; 103: 1644.