Kam směřuje farmakoterapie kardiovaskulárních chorob?
Polovina populace žijící v industriálních zemích umírá na kardiovaskulární onemocnění a téměř polovina prostředků spotřebovaných ve zdravotnictví se v těchto zemích vynakládá na léčbu onemocnění srdce a cév. Tyto dvě skutečnosti vedou farmaceutické společnosti k investování značných částek do vývoje nových léčebných modalit. V porovnání s posledním desetiletím minulého století, kdy byly v kardiologii prezentovány zásadní poznatky, často zcela měnící léčebné strategie, jsou výsledky klinických studií v posledních letech mnohem skromnější. V některých oblastech vývoj zcela stagnoval, takovým příkladem je léčba dyslipidémií, jinde, například v oblasti antitrombotik, slibně pokračuje.
Léčba kardiovaskulárních chorob má v řadě ohledů výjimečné postavení. Důvodů je několik. Předně, polovina populace v industriální společnosti umírá na kardiovaskulární onemocnění, a v důsledku toho se na léčbu onemocnění srdce a cév vynakládá téměř polovina prostředků spotřebovaných ve zdravotnictví. Tyto dvě skutečnosti vedou farmaceutické společnosti k investování enormních částek do vývoje nových léčebných modalit.
Vedle dostatečně silného ekonomického zázemí se pokrok v léčbě kardiovaskulárních chorob opíral o několik pilířů. Prvým bylo osvojení si principů medicíny založené na důkazech, což vedlo u nově zaváděných léčiv k jasnému určení hodnoty léčiva a jeho indikací. Vedle dodržování zásad „evidence based medicine" udělala kardiovaskulární farmakologie pokrok díky zásadním objevům v oblasti molekulární kardiologie. Další obor, který se v kardiologii prosazuje a bez jehož důsledného zavedení do praxe se neobejdeme, je farmakogenetika. Největší pokrok bylo možno pozorovat na přelomu tisíciletí, poslední léta, jak uvidíme, jsou „hubenější". Oblasti stojící na okraji vlastní farmakologie, tj. např. terapeutická angiogeneze, genový transfer nebo implantace zárodečných buněk, můžeme zmínit, tyto postupy se však orientaci článku vymykají. Lze jen konstatovat, že i zde vývoj jaksi přešlapuje a teprve hledá schůdnou cestu, jak se prosadit.
Ve srovnání se stavem před deseti lety, kdy jsme byli svědky exploze úspěšných studií hodnotících efekt nových léčiv v řadě kardiovaskulárních indikací a kdy jsme si ze všech „hot lines" sekcí kardiologických kongresů odnášeli zásadní poznatky, často zcela měnící léčebné strategie, jsou výsledky klinických studií v posledních letech výrazně skromnější. Budeme-li pátrat po příčině, zjistíme, že od počátku tisíciletí bylo v kardiologii zavedeno výrazně méně zcela inovativních léčiv. Jisté je, že předpisy regulačních orgánů a vysoké náklady na vývoj nového léku významně zpomalují inovaci. Navíc se setkáváme se stahováním perspektivních přípravků z trhu z důvodů obav firmy ze soudních sporů. Příkladem takového, z pohledu klinika nevýhodného zákroku je zastavení používání ximelagatranu z bezpečnostních důvodů v loňském roce. Srovnáme-li právě bezpečnostní profil ximelagatranu a jediného konkurenčního perorálně účinného antikoagulancia – warfarinu, musíme zastavení používání tohoto výrazně inovativního léčiva s širokým terapeutickým oknem jistě želet. Shrneme-li, pak je pravděpodobné, že takové úspěchy farmakoterapie kardiovaskulárních chorob, jichž jsme byli svědky v období devadesátých let, se již nebudou opakovat.
Zavádění farmakogenetických principů do klinické praxe
Než přistoupíme k přehledu, co nového se očekává v jednotlivých oblastech kardiologie, podívejme se blíže, proč je tak důležité zavádění farmakogenetických přístupů a proč právě v kardiologii je tato problematika aktuální. Jedním z hlavních úkolů farmakogenetiky je genotypizace enzymů metabolizujících a distribuujících léčiva v organismu. Rychlost odbourávání léčiva hraje totiž rozhodující úlohu v jeho expozici v organismu. U řady metabolických systémů jsou známy výrazné odchylky v aktivitě, rozdíly v rychlosti degradace mohou být až třicetinásobné. Důvodem těchto rozdílů je polymorfismus, tedy odlišné složení aminokyselin v proteinech enzymatických, transportních, regulačních či receptorových systémů. Tyto rozdíly stojí v pozadí za selháním efektu léku a zejména za vznikem nežádoucích účinků. Přitom nežádoucí účinky jsou jednou z nejčastějších příčin úmrtí, v USA jsou na 4. místě a ve Skandinávii byly předběhnuty sebevraždami. U léčiv metabolizovaných enzymy nezatíženými polymorfismem jsou nežádoucí účinky vzácnější. Můžeme tedy uzavřít, že právě interindividuální odchylky ve farmakokinetice léčiv mají zásadní klinický význam pro bezpečnost léčby.
Organismus má k dispozici v zásadě tři řady systémů, které zajišťují osud léku či, obecně řečeno, cizorodé látky (xenobiotika) v organismu. Jejich zapojení má zpravidla jediný cíl: usnadnit eliminaci xenobiotika z organismu, tj. zvýšit jeho rozpustnost ve vodě, aby mohlo být ledvinami vyloučeno. Z praktických důvodů dělíme tyto systémy podle typu reakcí, které zajišťují. Enzymy I. fáze mění charakter molekuly oxidací, resp. hydroxylací, enzymy II. fáze navazují vnesením hydrofilní skupiny, nejčastěji glukuronové či acetylové. Konečně ve III. fázi je xenobiotikum vyloučeno specifickými transportními systémy z organismu do moče, žluče či přímo sliznicí do střeva. Pořadí, v jakém je léčivo systémy biodegradováno a vyloučeno, může být různé a všechny cesty nemusí být užity.
Jak polymorfismus ovlivní toxicitu léčiva, možno demonstrovat na příkladu, jakým je léčivo s úzkou terapeutickou šíří – digoxin. Ten je substrátem glykoproteinu P. Nositelé genotypu TT s afunkčním glykoproteinem P mají zvýšené vstřebávání digoxinu a nižší clearance. Proto při běžném dávkování u nich mohou plazmatické hladiny digoxinu dosahovat trojnásobku běžných plazmatických hodnot a jejich koncentrace snadno dosahují toxického pásma se všemi průvodními jevy, jakými jsou výskyt arytmií a riziko náhlé smrti. Jak vyplynulo z reanalýzy studie DIG, tento fenomén vedl ke zvýšenému počtu náhlých arytmických smrtí v léčené skupině.
Komplikovanější situace je u jiného léčiva s úzkým terapeutickým oknem, u warfarinu. Dikumaroly inhibují účinek reduktázy vitaminu K, která vitamin K regeneruje a umožňuje aktivaci koagulačních faktorů. Pro nemocné s polymorfismem této reduktázy, který je přítomen asi u čtvrtiny naší populace, je typická její nízká aktivita, a z toho vyplývající výrazná senzitivita k působení warfarinu. Aby byla situace ještě složitější, za biodegradaci warfarinu je zodpovědný izoenzym CYP2C9. I tato oxidáza je pověstná svým polymorfismem. V populaci jsou tak rychlí, střední a pomalí metabolizátoři. Sejde-li se genetická výbava pro senzitivitu k warfarinu s jeho pomalým metabolismem, výsledná terapeutická dávka warfarinu se bude pohybovat kolem 1 mg a při užití běžného iniciačního dávkování 5 mg denně se nejspíše dočkáme krvácivé komplikace.
Posledním příkladem, kdy farmakogenetika rozhoduje o účinku léčiva, jsou lipofilní b-blokátory. Tato skupina léčiv, kam patří například metoprolol či karvedilol, je biodegradována velmi polymorfní oxidázou CYP2D6. U rychlých metabolizátorů je léčivo biodegradováno během 2–3 hodin, naopak u pomalých zůstává hladina i po 12 hodinách ještě v terapeutickém rozmezí. Interindividuální rozdíly expozice léčivu jsou tak až o řád rozdílné.
V současné době řada oddělení klinické farmakologie již vyšetření základního spektra polymorfismů metabolických a transportních systémů rutinně provádí, a dokonce řada vyšetření je zařazena do systému úhrad. Vývoj spěje k tomu, že po narození bude izolována z pupečníkové krve DNA a jednotlivé relevantní polymorfismy zvyšující riziko určitých chorob a ovlivňující farmakokinetiku či farmakodynamiku léčiv budou stanoveny. Na základě genotypizace pak budeme moci účinněji a zejména bezpečněji léčit.
Důslednější sledování rizika závažných lékových interakcí
Dalším velmi důležitým trendem, který zlepší kvalitu a bezpečnost farmakoterapie, je důsledné sledování rizika vzniku závažných lékových interakcí. Na základě desítek prospektivních studií a pacientských registrů v USA či ve Skandinávii je doloženo, že ve vyspělých populacích se objevuje asi 400 úmrtí na každý milion obyvatel, která jdou na vrub nežádoucích účinků léčiv. Na větší části těchto úmrtí, tj. asi na 70 %, se spolupodílejí lékové interakce. Ač se u nás nežádoucí účinky hlásí jen vzácně, dá se analogicky odvodit, že téměř tři tisíce nemocných v České republice z důvodů lékových interakcí ročně zemře. Číslo bude pravděpodobně ještě vyšší, neboť jsme národ farmakofágů, na hlavu zkonzumujeme průměrně 32 balení léků ročně a řadíme se tím po Francii a Maďarsku k evropské špičce.
Riziko fatálních interakcí je významné zejména při komedikaci dvou léčiv, z nichž jedno je inhibitorem systému a druhé substrátem některého z metabolických systémů (oxidáz skupiny izoenzymů CYP, transferáz či membránových transportních proteinů). Inhibice společného systému vede k toxické kumulaci léčiva a k riziku nežádoucího účinku. Příkladem takovéto interakce byl efekt blokátoru kalciového kanálu mibefradilu inhibujícího CYP3A4, který metabolizuje např. simvastatin. Série úmrtí po léčbě touto kombinací léčiv vedla ke stažení mibefradilu z trhu. Zdaleka ne všechny inhibitory metabolických systémů je však možno vyloučit z terapie. Těžko bychom si mohli představit v současné době arytmologii bez amiodaronu. Toto antiarytmikum je bohužel výrazným inhibitorem téměř všech významných metabolických systémů: CYP3A4, CYP2C9, CYP2D6 či glykoproteinu P. Se zvýšením hladiny warfarinu a se vzestupem INR při komedikaci s amiodaronem se setkal každý kardiolog. Ne každý si však vzestup INR správně vysvětlil. Přitom tato kombinace léčiv je častá například u pacientů s fibrilacemi síní, z databází zdravotních pojišťoven vyplývá, že se jedná o jednu z nejčastějších závažných lékových interakcí u nás.
Podobným příkladem je sildenafil. Každý z nás ví, že současné podání nitrátů vede k inhibici degradace NO a k následné masivní vazodilataci s rizikem šoku. Sildenafil, který působí řádově několik hodin, je substrátem CYP3A4. Když zablokujeme účinek této oxidázy například verapamilem, diltiazemem, makrolidovými antibiotiky, řadou psychofarmak či flavonoidy z řady ovocných šťáv, prodloužíme efekt sildenafilu několikanásobně. Vedle toho, že docílíme „víkendového potěšení", musíme ale očekávat, že ještě druhý den je aktuální riziko interakce. Z těchto důvodů jde na vrub komedikace sildenafilu a nitrátů do dnešní doby více než stovka úmrtí.
Interakce nemusejí mít vždy charakter zvýšení účinku léčiv, naopak relativně často se setkáme i se selháním jejich efektu. Že takováto interakce může být rovněž významná, můžeme demonstrovat na příkladu klopidogrelu, který podáváme ve formě proléčiva (prodrug). K jeho konverzi na aktivní metabolit je nutná funkční oxidáza CYP3A4. Při komedikaci s inhibitory tohoto izoenzymu dochází k selhání protidestičkového účinku. Vedle výše uvedených inhibitorů je např. dokumentována ztráta efektu klopidogrelu po komedikaci s antidepresivy typu SSRI, s antimykotiky či po grapefruitové šťávě.
Podobných závažných interakcí je v medicíně příliš mnoho – téměř tři tisíce charakteru kontraindikací a pět tisíc interakcí velmi závažných. Proto si je nikdo z nás nemůže zapamatovat a budeme nuceni k daleko užší spolupráci s automatickými informačními systémy. Podmínkou bude rutinní zavádění elektronické zdravotní dokumentace (optimálně obsahující i pacientův genotyp) s přehledem medikace od různých lékařů. Tato informace v kombinaci s počítačovou databází lékových interakcí pak na riziko závažné interakce upozorní předepisujícího lékaře, případně vydávajícího lékárníka. Obě podmínky již jsou splněny, jsou k dispozici zdravotní knížky typu IZIP i spolehlivá databáze lékových interakcí. Bude jen na nás, kdy informace propojíme. V zájmu našich nemocných bychom neměli otálet.
Jaké inovace můžeme očekávat v jednotlivých oblastech?
Po tomto obecnějším pohledu, který nastínil, jaké úkoly stojí před námi a kde se situace výrazně mění, přistupme k vlastnímu přehledu, co je nového. Nelze probrat vše nové v jednotlivých oblastech kardiologie, to je nad možnosti tohoto článku. Proto byly vybrány dvě oblasti, kde se v posledních letech nejvíce změnilo a kde se další vývoj očekává: léčba dyslipidémií a problematika antitrombotik.
Jaké jsou pokroky v léčbě dyslipidémií?
Léčba dyslipidémií patří mezi největší úspěchy medicíny dvou posledních desetiletí. Zásah do metabolismu lipidů pomáhá zvládat pandemii chorob na podkladě aterotrombózy. Vedle trendu ke snižování cílových hodnot aterogenních lipidů s následným zvyšováním terapeutických dávek statinů spějeme ke kombinační léčbě. Příkladem je kombinace statinů s blokátorem resorpce cholesterolu ezetimibem při vysokých hladinách LDL, eventuálně kombinace statinů s fibráty či s niacinem při nízkých hodnotách HDL či při smíšené dyslipidémii.
Farmakoterapie zvýšených hladin LDL cholesterolu využívá více strategických přístupů. Vychází z fyziologických poznatků, že hladina cholesterolu v hepatocytech je v rovnovážném stavu udržována syntézou cholesterolu (cestou hydroxy-methylgluta-ryl-koenzym A reduktázy), vychytáváním cholesterolu receptory LDL, skladováním ve formě esterů (cestou acetylkoenzym A-cholesterolacyltransferázy – ACAT) a sekrecí cholesterolu do žluče jako žlučových kyselin či přímo jako cholesterolu. Plazmatická hladina LDL je kontrolována jak syntézou, tak zejména aktivitou receptorů LDL v játrech. Pokles intracelulární hladiny cholesterolu vede k expresi receptorů LDL, obnoví se koncentrace cholesterolu v buňce a klesá hladina LDL v plazmě.
Ve fázi syntézy cholesterolu byly testovány různé látky inhibující steroidogenezi na různých úrovních. Ukázalo se, že je nutno zabrzdit syntézu již v časných fázích tak, aby nedošlo k nežádoucímu hromadění prekurzoru. V praxi jsou proto užívány pouze inhibitory HMG-CoA reduktázy, statiny. Vývoj inhibitorů skvalensyntázy, která blokuje steroidogenezi ve vyšších úrovních, zatím nepřekročil fázi preklinických studií.
Inhibice resorpce cholesterolu
V popředí zájmu jsou postupy inhibující resorpci cholesterolu na úrovni enterocytu (obr. 1).
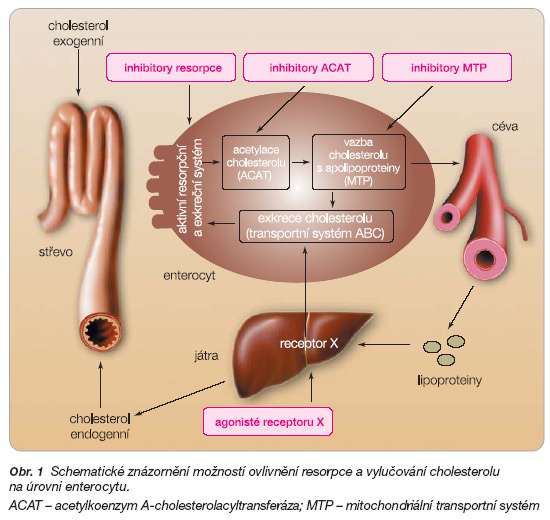
Ezetimib, blokátor transportního systému (Niemann-Pick C1-like 1 protein) umožňujícího vstřebávání cholesterolu na úrovni povrchu buněk střevních klků, užíváme již léta a je často podáván u závažných dyslipidémií. Dosud byly předloženy jen studie dokumentující efekt na lipidové spektrum v monoterapii nebo v kombinaci se statiny, fibráty či pryskyřicemi. Proto s napětím očekáváme výsledky prognosticky zaměřených klinických studií.
Vedle blokády na úrovni Niemannova-Pickova proteinu jsou patrné snahy zabránit vstřebání cholesterolu i na vyšších etážích. Volný cholesterol v enterocytu může být intracelulárním transportním mechanismem (protein ABC) vyloučen zpět do střeva či je esterifikován pomocí ACAT. Estery cholesterolu jsou opět specifickým proteinem (mitochondriální transportní systém – MTP) navázány na apolipoproteiny a v chylomikronech transportovány do jater.
Dalším krokem, kde lze zamezit vstřebávání cholesterolu v enterocytu, se zdála být blokáda esterifikace inhibitory ACAT. Tato cesta měla v experimentu, vedle poklesu hladiny LDL cholestrolu, i další přímý antiaterogenní efekt. Cholesterol je totiž akumulován v makrofázích jako ester. Nadbytek tohoto cholesterol oleátu vede k tvorbě pěnových buněk na jedné straně a na straně druhé k aktivaci makrofágů s uvolněním řady cytokinů, metaloproteináz, oxidačních a lipolytických enzymů, které potencují aterogenezi, zvyšují riziko destabilizace plátu a potencují trombotickou pohotovost. U obou inhibitorů ACAT – avasimibu i pactimibu – bylo bohužel předčasně ukončeno klinické hodnocení. Ani u jednoho se nepodařilo prokázat regresi koronárních plátů pomocí intrakoronárního ultrazvuku (IVUS), naopak proti placebu byl patrný spíše proaterogenní efekt. Podobně nebyl prokazatelný efekt na prodloužení klaudikačního intervalu. Je tedy pravděpodobné, že se vývoj této skupiny ukončil.
Další možností, jak snížit resorpci cholesterolu a triglyceridů, je inhibice MTP. Bohužel, studie s představitelem inhibitorů MTP imlitapidem nepokročily během posledních let do fáze klinického zkoušení a je otázkou, zda vývoj této skupiny rovněž nebyl zastaven.
Zajímavou a slibnou možností, jak snížit hladinu LDL cholesterolu, vedle snížení syntézy a snížení resorpce, je zvýšit jeho odpad do střeva. Dosavadní způsob vychytávání cholesterolu neresorbovatelnými pryskyřicemi není optimální pro jeho horší snášenlivost. Proto je pozornost soustředěna na enterocytální transportní systém (ABC-A1), který zajišťuje vylučování cholesterolu do lumina střeva a snižuje vstřebávání cholesterolu. Exprese systému ABC-A1 je pod kontrolou jaterních receptorů X (LXR). Agonisté těchto receptorů jsou vyvíjeny řadou firem, nadějné jsou například diterpeny odvozené od kyseliny akantové či deriváty chinolonů. I když je řada prací dokumentujících příznivý dopad stimulace LXR na lipidové spektrum, zánětlivou odpověď i na antiaterogenní efekt v experimentu, nejsou žádné zprávy o tom, že by bylo započato s klinickým hodnocením.
Postupy zvyšující hladinu HDL
Nejméně stejně důležitým postupem, jak bojovat s aterogenezí, je – vedle snížení hladiny LDL cholesterolu – zvýšení hladiny lipoproteinových částic s vysokou denzitou. HDL cholesterol je zcela nezávislý faktor inverzně korelující s rizikem aterotrombotických příhod. Je-li LDL cholesterol spojen s vývojem endoteliální dysfunkce, s aktivací vazoadhezních molekul, s navozením prokoagulačního stavu a s akumulací lipidů v jádru aterosklerotického plátu, pak HDL má funkci právě opačnou. Asi nejdůležitější je mobilizace nadbytečného cholesterolu z periferních tkání do jater a jeho vyloučení do žluči.
Nejdéle používanou skupinou k léčbě dyslipidémií, zejména k léčbě hypertriglyceridémie a k léčbě snížené hladiny HDL cholesterolu, jsou deriváty kyseliny fibrové, fibráty. Mechanismus účinku představuje stimulace specifických receptorů řídících metabolismus lipidů a glycidů v buněčných organelách – peroxizomech. V současné době je patrný jednoznačný ústup od užívání fibrátů, který pramení z rozpačitých výsledků klinických studií, příkladem je studie FIELD. Zda se v budoucnu objeví nové molekuly v této jinak teoreticky velmi zajímavé skupině, je otázkou. Většina projektů se spíše soustřeďuje na peroxizomální receptory – PPAR-g, resp. PPAR-a, které se uplatňují v lipidovém i glycidovém metabolismu. Jejich stimulace v adipocytech např. antidiabetiky glitazonové řady (thiazolidindiony) zvyšuje citlivost k inzulinu či duální stimulace některými fibráty má komplexní efekt na lipidový i glycidový metabolismus.
Vzestup hladiny HDL cholesterolu je dokumentován po podávání kyseliny nikotinové, niacinu. Mechanismus působení tohoto léčiva a dalších léčiv odvozených od kyseliny nikotinové spočívá v uvolnění volných mastných kyselin v periferních tkáních, jejich vychytání v játrech, v následném snížení syntézy lipoproteinů bohatých na triglyceridy a v útlumu konverze VLDL na LDL. Léčba niacinem byla provázena významným poklesem kardiovaskulární morbidity i mortality (Coronary Drug Project) či regresí koronární aterosklerózy (CLAS, FATS). Retardovaný niacin, který je v registraci, by měl být v tomto či nejpozději v příštím roce zaveden do klinické praxe i v naší republice.
Poněkud déle asi budeme muset čekat na zavedení léčby homologním HDL. Preklinické studie s infuzemi rekombinantním HDL byly příznivé. Regrese depozit cholesterolu a jeho esterů z aterosklerotických plátů, tzv. HDL delipidizace, byla velmi rychlá. Klinické zkušenosti byly bohužel jiné. Komplex apolipoproteinu A1 s fosfatidylcholinem (CSL-111), tj. analogu HDL partikulí, byl testován v rámci II. fáze hodnocení, nebyla pozorována změna složení koronárních plátů při vyšetření IVUS a studie byla předčasně ukončena pro vzestup hladin jaterních testů již při krátkodobé léčbě. Předpoklad, že by po koronární příhodě či při známkách nestability bylo možno započít s infuzemi rychle působícího rekombinantního homologního HDL (či jeho analog) a po stabilizaci stavu pokračovat v klasické léčbě dyslipidémie, dostal vážnou trhlinu.
Nejdále pokročily klinické studie u nové skupiny inhibitorů CETP (cholesterol ester transfer protein). Tento protein urychluje přenos esterů cholesterolu z HDL do aterogenních částic LDL a VLDL. Dá se tedy říci, že dochází ke zpomalení clearance HDL a částice naplněné cholesterolem odčerpaným z periferních tkání se hromadí, a zvyšují tak hladinu HDL. Podávání inhibitorů CETP vedlo v experimentu i při klinickém hodnocení k nevídanému zvýšení hladin HDL cholesterolu (téměř ke zdvojnásobení) a ke snížení hladin LDL cholesterolu. Efekt byl patrný jak v monoterapii, tak i v kombinaci se statinem. Blokáda CETP prvým představitelem této skupiny, torcetrapibem, která v experimentu vedla k antiaterogennímu efektu, však v klinických studiích selhala. Po počátečním zklamání, kdy se nepodařilo podáváním kombinace statinu a torcetrapibu navodit regresi koronárních plátů (srovnání s placebem), byla velká mortalitně-morbiditní studie ILLUMINATE předčasně ukončena pro zvýšení úmrtnosti v aktivní větvi. Hypotetických důvodů, proč k tomu došlo, je více. Jedním z nejpřijatelnějších je, že zpomalení koloběhu HDL cholesterolu sníží reverzní transport cholesterolu z periferních tkání, a je tak navozen vlastní proaterogenní efekt, který neutralizuje příznivý účinek statinu. Pro tuto teorii by mohl svědčit výsledek studie ILLUSTRATE, která neprokázala regresi ateromových plátů při vyšetření IVUS. Druhým, rovněž přijatelným vysvětlením je, že zásah do metabolismu HDL inhibuje NO syntázu s navozením endoteliální dysfunkce a s navazující vazokonstrikcí, trombogenezí a aterogenezí. Pro tuto teorii svědčí významný vzestup krevního tlaku, který je v torcetrapibových větvích opakovaně pozorován. Nicméně vzhledem k tomu, že není jasné, zda nepříznivý klinický efekt není specificky vázán na vlastní molekulu, vývoj ve skupině není ukončen. Jsou zprávy o připravovaném klinickém hodnocení jiného inhibitoru CETP.
Shrneme-li současné trendy v léčbě dyslipidémií, je patrný posun intervence LDL cholesterolu ve smyslu „čím níže, tím lépe" s užitím především kombinace nižších dávek hypolipidemik působících na různých úrovních (např. blokáda syntézy statinem s inhibicí resorpce ezetimibem). V oblasti nových molekul určených k léčbě dyslipidémií nebyl v posledních letech zaznamenám větší pokrok, naopak jsme svědky spíše zklamání. Minimálně dvě nejnadějnější skupiny – inhibitory ACAT a inhibitory CETP – nesplnily vkládané naděje. Otázkou je též, zda bude pokračovat vývoj v oblasti substituce homologním HDL.
Jaké jsou pokroky v léčbě trombotických stavů?
Trombotické komplikace aterogeneze jsou nejčastější příčinou úmrtí v naší civilizaci, vlastní ateroskleróza by bez nasedající trombózy byla jen relativně benigní chorobou, neboť to, co nemocného zabíjí, je akutní trombotický uzávěr. Zavedení protidestičkové či alternativní antikoagulační léčby snížilo mortalitu nemocných po překonaném infarktu o 20–30 %, antikoagulační a protidestičková léčba snížila úmrtnost při akutních koronárních příhodách ještě výrazněji. Není proto divu, že inovace v oblasti léčby trombogenních stavů zajímá nejen klinika, ale zejména vývojová oddělení farmaceutických firem.
Obecně je možno říci, že trendem v antitrombotické léčbě je kombinace protidestičkových léčiv (působících na různé aktivující receptory nebo blokující aktivaci a agregaci) nebo kombinace protidestičkových léčiv s antikoagulancii (např. kombinace blokátorů aktivace s trombinovými inhibitory či s antagonisty vitaminu K). Vzhledem k tomu, že kombinační léčba přináší větší riziko krvácivých komplikací, které je proti monoterapii zvýšené o absolutní 1 % během roku léčby, je kombinační léčba vyhrazena pro nemocné s vyšším rizikem, tj. například u akutních koronárních příhod a v časném období po jejich překonání.
V hemostáze vidíme prolínání primární hemostázy s vlastní koagulací. Vytvoření primárního trombu akceleruje koagulační kaskádu, naopak uvolnění malého množství trombinu aktivuje trombocyty. Proto je dělení na protidestičková léčiva a na antikoagulancia do jisté míry umělé, nicméně je výhodné z didaktických důvodů.
Vývoj v oblasti protidestičkové léčby
V procesu formace destičkového trombu jsou klíčové čtyři základní kroky: adheze, aktivace, degranulace (sekrece) a agregace (obr. 2).
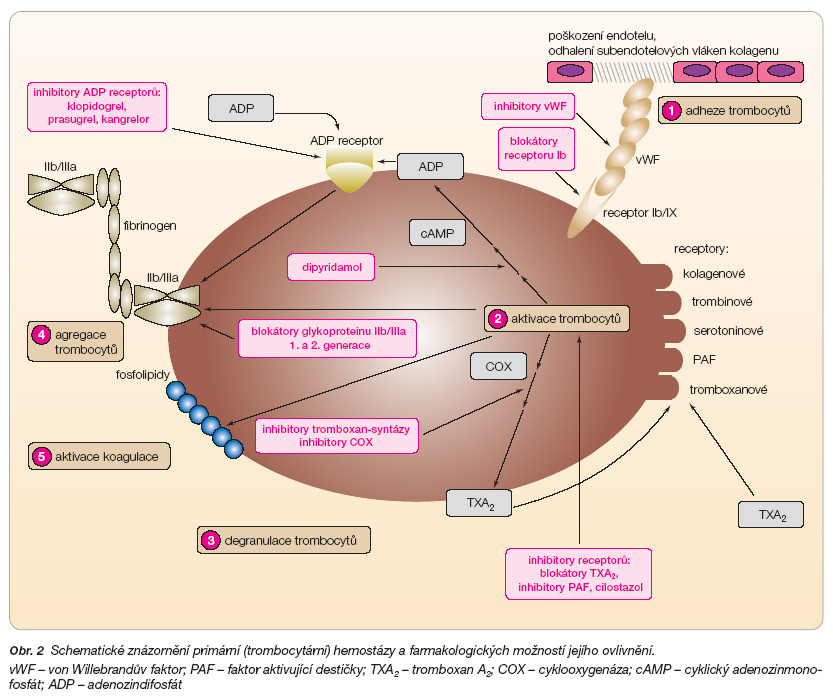
Současná farmakologie nám umožňuje blokovat účinek trombocytů na úrovni aktivace (např. inhibicí cyklooxygenázy či blokátory receptorů pro ADP) a agregace (inhibitory receptorů IIb/IIIa). Přitom zabránění adhezi trombocytu na obnažená kolagenní vlákna by se logicky nabízelo jako nejvýhodnější. Ve fázi klinického zkoušení jsou blokátory adheze trombocytů na bázi protilátek, směřované zejména proti trombocytárním receptorům Ib a IIa nebo proti bivalentnímu vazebnému proteinu – vWF (von Willebrandův faktor). Vzhledem k tomu, že v posledních letech nejsou žádné zprávy o iniciaci klinického hodnocení, je nepravděpodobné, že by vývoj v této oblasti výrazněji pokročil. Jedinou výjimkou je duální inhibitor kamonagrel, který v nízké koncentraci inhibuje adhezívní receptory a ve vyšší též tromboxansyntázu. Zatím není zpráv, zda bylo započato s klinickým hodnocením.
Zabránění aktivaci trombocytu patří v současné době ke zlatému standardu protidestičkové léčby. Podávání acetylsalicylové kyseliny (ASA) inhibující tromboxanovou cestu aktivace či tiklopidinu nebo klopidogrelu příznivě ovlivňuje morbiditu a mortalitu řadu let. Snahy o blokádu serotoninových receptorů se příliš mnoho neosvědčily, naftidrofuryl má protidestičkový efekt malý, výjimkou je pouze cilostazol, který je v řadě zemí s výhodou užíván k léčbě ischemické choroby dolních končetin.
Novinek ve skupině inhibitorů syntézy tromboxanu A2 (TXA2) či blokátorů jeho receptorů není mnoho, alternativou ASA je triflusal, rovněž nevratně inhibující biosyntézu tromboxanu. Jeho efekt v profylaxi recidivy mozkové příhody či infarktu myokardu je srovnatelný s ASA, v kombinaci s antikoagulační léčbou byl účinnější v profylaxi tromboembolie u nemocných s fibrilací síní než antikoagulace samotná. Je tedy pravděpodobné, že se triflusal stane alternativou ASA u nemocných s její intolerancí či při rezistenci k léčbě. Úspěšnému zavedení triflusalu do klinické praxe zatím brání vysoká cena.
Jinak vývoj v oblasti blokády syntézy TXA2 a blokády jeho receptorů stagnuje. Inhibitory tromboxanových receptorů (ridogrel či nidrogrel) i inhibitory tromboxansyntázy (dazoxiben a ozagrel) se neosvědčily, léčba byla zatížena větším rizikem krvácení při srovnatelném efektu v porovnání s ASA. Anagrelid, blokující trombocyty již ve fázi megakaryocytu, v profylaxi trombotických komplikací rovněž nepředčil standardní léčbu. Jeho jedinou indikací je nekontrolovaná megakaryocytopoéza u esenciální trombocytózy. Podobně zklamal vývoj v oblasti inhibitorů fosfolipázy A2 (PLA2), enzymu uvolňujícího kyselinu arachidonovou z fosfolipidů buněčných membrán. Uvolnění kyseliny arachidonové je nutným krokem k syntéze eikosanoidů, a tedy i tromboxanu A2. Po fázi nadějných laboratorních výsledků se vývoj zastavil již v preklinické fázi.
Zklamaly-li inovativní léčebné postupy v oblasti aktivace destičky tromboxanovou cestou, pak v oblasti blokády aktivace cestou ADP se rýsují nejméně dvě slibné molekuly. Na povrchu trombocytu jsou tři různé receptory pro ADP, významné jsou receptory P2Y1 a P2Y12. Oba se účastní aktivace a potencují agregaci, receptor P2Y1 mobilizuje ionizované kalcium ze zásobních vezikul a stimuluje časné fáze agregace, receptor P2Y12 naopak aktivuje destičky inhibicí adenylátcyklázy a potencuje pozdní fáze agregace. Thienopyridiny (klopidogrel a tiklopidin) inhibují agregaci zprostředkovanou receptory P2Y12. Jejich význam v léčbě nestabilních forem ischemické choroby srdeční a při koronárních intervencích je nepochybný a kombinace s ASA je považována za zlatý standard. Naopak v indikaci léčby méně rizikových stavů, tj. v primární prevenci či u stabilizovaných nemocných v prevenci sekundární, se kombinace příliš neosvědčila. Riziko krvácení při duální protidestičkové inhibici může převýšit vlastní přínos antitrombotické léčby. Před ukončením III. fáze klinického hodnocení je prasugrel, představitel druhé generace thienopyridinů. Kangrelor, non-thienopyridinový blokátor ADP receptorů P2Y1 i P2Y12, do III. fáze klinického hodnocení nedávno vstoupil.
Efekt prasugrelu, ireverzibilního blokátoru receptorů P2Y12, je, stejně jako u klopidogrelu, zprostředkován aktivním metabolitem. Oproti klopidogrelu však má řadu výhod, ve farmakoekvivalentních dávkách je patrný výraznější efekt na aktivaci trombocytů. Při perorálním podání je nástup účinku patrný již za 30 minut a efekt trvá 72 hodin a je dokumentována méně častá rezistence než při léčbě klopidogrelem. V souhlase s větším efektem je i výskyt krvácivých příhod vyšší, resp. tyto byly protrahovanější. Ve studii JUMBO u nemocných po koronární intervenci byl pozorován trend k poklesu větších kardiovaskulárních příhod ve srovnání s léčbou klopidogrelem. Větší studie (TRITON), která by měla prokázat „superioritu", probíhá.
Velmi slibné výsledky v preklinické fázi testování a v časných fázích klinického hodnocení byly zaznamenány u kangreloru, non-thienopyridinového reverzibilního inhibitoru receptorů P2Y12 a P2Y1. Jeho předností je velmi rychlý nástup účinku (v řádu minut), krátký účinek (t1/2 je jen 3–5 minut) a nepřítomnost aktivního metabolitu (nehrozí tedy riziko blokády biokonverze). Díky tomu, že efekt je zprostředkován převážně blokádou receptorů P2Y1, dochází k aditivnímu účinku při komedikaci s klopidogrelem. Vzhledem ke krátkému efektu a nutnosti kontinuální parenterální aplikace je testován u akutních koronárních příhod.
Agregace krevních destiček je vyvrcholením primární hemostázy. Během aktivace trombocytu dochází ke změně konfigurace povrchového transmembránového receptoru GP IIb/IIIa, při které dojde k odhalení vazebných míst bivalentních proteinů, které váží aktivované trombocyty navzájem (tzv. aktivace receptoru). K inhibici agregace je nutno inhibovat asi 80 % těchto receptorů, kterých je na povrchu trombocytu asi 60–100 tisíc. U akutních koronárních syndromů se osvědčily parenterálně účinné blokátory GP IIb/IIIa. Perorální blokátory, tzv. fibany, se ukázaly být krátkodobě účinné a objevoval se rebound fenomén s hyperagregačním stavem. Léčba byla navíc zatížena vysokým výskytem krvácivých příhod.
Bohužel, v zavádění nové generace inhibitorů GP IIb/IIIa s delší dobou účinku a pevnější vazbou k receptoru není velký pokrok. Po zveřejnění výsledků studie s prvním představitelem další generace blokátorů – lotrafibanem – u nemocných s chronickými formami aterosklerotického postižení (s převahou mozkové aterosklerózy) se vývoj v této skupině výrazně zpomalil. Tato prognostická studie totiž dokumentovala vzestup mortality i krvácivých komplikací v aktivní větvi léčby.
Vývoj v oblasti antikoagulační léčby
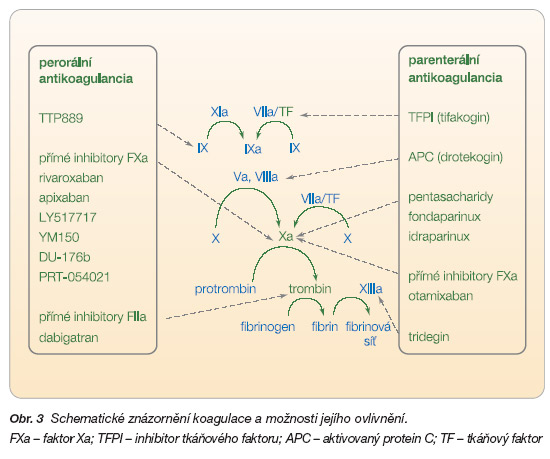
Sekundární hemostáza neboli vytvoření stabilní fibrinové sítě je zprostředkováno soustavou proteáz, jejímž cílem je postupně autokatalýzou zvyšovat svůj účinek (amplifikace). Konečným cílem je aktivovat poslední proteázu řetězce – trombin, který aktivuje polymerizaci rozpustného fibrinogenu na nerozpustnou fibrinovou síť (obr. 3). Stabilita této sítě je zajištěna vzájemným navázáním jednotlivých vláken polymeru fibrinu. To je katalyzováno faktorem XIIIa. Tento terminální krok pak činí sekundární trombus odolnější k fibrinolýze plazminem. V centru celé koagulační kaskády je trombin, klasifikace antitrombotik je proto založena na jejich vztahu k trombinu. V iniciálních stadiích je důležité uvolnění tkáňového faktoru, který iniciuje uvolnění malého množství trombinu, a tento krok pak celou koagulační kaskádu nastartuje. Aktivita trombinu je hlídána účinným regulačním systémem: antitrombinem, který vazbou trombinu inaktivuje. Afinita samotného antitrombinu k vazebnému místu trombinu je malá, teprve v komplexu s heparanem či s hepariny se celá reakce rozbíhá.
Na rozdíl od protidestičkových léčiv, kde máme alespoň dvě vyhovující léčiva – acetylsalicylovou kyselinu a klopidogrel, je u antikoagulancií situace problematičtější. Máme účinná a spolehlivá léčiva pro krátkodobou léčbu – nízkomolekulární hepariny či pentasacharidy, problém však je s léčbou dlouhodobou. Antikoagulancia, která v současné době užíváme v léčbě chronických stavů, mají mnoho nevýhod. Jediné perorálně dostupné antikoagulans – warfarin – je často uváděným příkladem velmi problematického léčiva. Účinná dávka je ovlivňována příjmem vitaminu K, polymorfismem cílového enzymu (reduktázy vitaminu K) rozhodujícího o citlivosti k warfarinu, polymorfismem CYP2C9, izoenzymu metabolizujícího warfarin nebo četnými interakcemi s léčivy na úrovni tohoto cytochromového systému. Vzhledem k úzkému terapeutickému oknu musí být dávka antivitaminu K velmi přesná, a proto se účinné dávky mohou u jednotlivých nemocných lišit o řád.
Nízkomolekulární hepariny, heparin či pentasacharidy mají nevýhodu parenterální aplikace. Proto je zřejmé, že potřebujeme nová antikoagulancia, která by byla výhodná pro dlouhodobé použití, tedy optimálně účinná perorálně.
Inhibitory trombinu
Jak již bylo řečeno, klíčovým místem koagulační kaskády je trombin. Ten je klasicky možno inhibovat aktivací antitrombinu např. hepariny. Nevýhodou této nepřímé inhibice je to, že nedocílíme inaktivace trombinu vázaného na trombin, proto se po uvolnění z této vazby koagulační kaskáda opět rozbíhá. Z těchto důvodů jsou perspektivnější přímé trombinové inhibitory, které jsou schopné inaktivovat i trombin již navázaný.
Přímé trombinové inhibitory, které inhibují trombin vazbou v oblasti aktivního místa trombinu, užíváme již od středověku. Při mozkové mrtvici se například přikládaly pijavice, z jejichž slin se po přisátí resorboval přímý inhibitor trombinu – hirudin. Hirudiny i jeho rekombinantní analoga či argatroban mají bohužel nevýhodu parenterální aplikace. Proto jejich indikací je dnes heparinem indukovaná trombocytopenie, ev. akutní koronární příhody. Prvý, již výše zmíněný perorální přímý inhibitor trombinu – ximelagatran – byl užíván v profylaxi tromboembolických příhod jen krátce. V únoru 2006 byl bohužel stažen z trhu z důvodů podezření na hepatotoxický účinek.
V současné době je ve III. fázi klinického hodnocení nový přímý reverzibilní inhibitor trombinu – dabigatran. Vzhledem k tomu, že se jedná o velmi nadějné léčivo, bude vhodné uvést základní fakta. Perorálně dostupné proléčivo (prodrug) dabigatran etexilát (BIBR 1048) je metabolizováno na aktivní substanci (BIBR 953 ZW) s rychlým nástupem účinku (maximální efekt již do hodiny) a s dlouhou dobou působení, která umožňuje podat léčivo ve dvou denních dávkách. Ve studii BISTRO II byl porovnáván dabigatran (podávaný v různých dávkách) s enoxaparinem v indikaci profylaxe tromboembolických komplikací pacientů podstupujících ortopedické operace. V dávce 2krát denně 150 mg byla léčba dabigatranem účinnější při srovnatelném výskytu větších krvácivých příhod. V současné době je dabigatran testován ve III. fázi klinického hodnocení v indikaci profylaxe tromboembolických příhod u pacientů s ortopedickými operacemi a dále u pacientů s fibrilacemi síní.
Z ostatních přímých inhibitorů trombinu není vývoj žádného tak daleko, jako je tomu v případě dabigatranu. Proslýchá se, je vyvíjen nástupce ximelagatranu bez negativního dopadu na jaterní enzymy, nicméně v této oblasti nebyly zatím uvolněny žádné oficiální informace. O výsledcích hodnocení žádného z tripeptidů kovalentně obsazujících aktivní místo trombinu (efegatranu, inogatranu či napsagatranu) nejsou v posledních letech žádné zprávy, je tedy pravděpodobné, že jejich vývoj byl zastaven. Podobně není patrný pokrok v oblasti vývoje perorálně účinných nepřímých inhibitorů trombinu. Nedostatek informací o pokrocích ve vývoji perorálních derivátů heparinu (tzv. rd-hepariny a rd-LMWH) nasvědčuje, že tato skupina dosud nepřekročila fázi preklinického hodnocení.
Inhibitory faktoru Xa
Faktor Xa (FXa) má výsadní postavení v katalýze konverze protrombinu na trombin. Aktivace FXa je společným uzlem „vnitřní" i „vnější" cesty koagulační kaskády. Cílení blokády na FXa má racionální podklad, inhibice tohoto faktoru by měla být výhodnější z několika důvodů. Faktor Xa není, na rozdíl od trombinu, zapojen do antitrombotického systému směřujícího k aktivaci proteinu C a S. Na základě studií in vitro i in vivo porovnávajících neselektivní (heparin a nízkomolekulární hepariny) a selektivní inhibitory FXa (pentasacharidy) se ukázalo, že selektivita inhibice FXa je výhodná, je přítomen větší antikoagulační potenciál, je přítomno širší terapeutické okno a není přítomen rebound fenomén. Přirozeným inhibitorem účinku faktoru Xa je (podobně jako u trombinu) antitrombin. Ten, spolu se svým kofaktorem heparanem či heparinem, tlumí aktivitu této proteázy nejen v rámci koagulace, ale též v produkci cytokinů. Hepariny působí nejen na inhibici trombinu, ale i na inhibici faktoru Xa. Pentasacharid fondaparinux, odvozený z heparinu, rovněž aktivuje antitrombin, ale vzniklý komplex působí specificky pouze na faktor Xa. Nevýhodou těchto nepřímých inhibitorů je nedostatečný efekt na inhibici FXa vázaného v protrombinázovém komplexu.
Významné pokroky lze pozorovat ve vývoji přímých inhibitorů faktoru Xa. Přímé inhibitory faktoru Xa, na rozdíl od nepřímých, nevyžadují ke svému působení „zprostředkovatelskou" molekulu a díky výrazně menší molekule nic nebrání inhibici faktoru Xa již zavzatého do protrombinázového komplexu. Neexistuje jiná oblast vývoje antikoagulační léčby, která by byla tak silně zastoupena jako skupina přímých inhibitorů FXa. Rivaroxaban i apixaban jsou ve III. fázi klinického hodnocení a pět dalších molekul, zatím jen s kódovým označením, vstoupilo do fáze II.
Z pohledu vhodnosti pro chronickou antikoagulační léčbu se zdá být nadějným kandidátem rivaroxaban (BAY 59-7939), přímý inhibitor FXa. Jeho předností je velká selektivita, prediktabilní efekt, účinek při perorální aplikaci a efekt trvající déle než 24 hodin, který umožňuje aplikaci jedenkrát denně. Výsledky II. fáze hodnocení dokládají antitrombotický efekt srovnatelný se standardní léčbou (enoxaparin + antivitamin K). V rozmezí dávky 5–20 mg denně byl jeho terapeutický i krvácivý potenciál v profylaxi tromboembolie u pacientů podstupujících ortopedickou operaci srovnatelný s enoxaparinem (pilotní studie RECORD). V indikaci léčby flebotrombóz byla dokumentována stejně velká regrese trombu i stejná incidence tromboembolizace jako u enoxaparinu v kombinaci s antivitaminem K (studie ODIXa-DVT). Pokračují rozsáhlé studie v oblasti profylaxe tromboembolizace v průběhu větších ortopedických výkonů (RECORD), v léčbě plicní embolie (EINSTEIN), v profylaxi tromboembolizace při fibrilaci síní (ROCKET AF) či v léčbě akutních koronárních příhod (ATLAS – TIMI 46).
Méně výsledků o efektu máme u jiného přímého inhibitoru FXa – apixabanu. Podobně jako rivaroxaban je perorálně účinný a je dokumentována selektivita k faktoru Xa. Hodnocení v indikaci léčby flebotrombóz, v prevenci plicní embolizace u onkologicky nemocných, v prevenci tromboembolizace u fibrilace síní či v léčbě akutních koronárních příhod probíhá (kolekce studií BOTTICELLI).
K parenterálně účinným přímým inhibitorům FXa patří otamixaban. Jeho efekt je rychlý, ale krátkodobý, proto je podáván ve formě iniciální dávky (bolus) a je pokračováno infuzemi. Jeho léčebný potenciál u akutních koronárních příhod je prověřován.
Z farmakologického hlediska i z dostupných výsledků klinického hodnocení je pravděpodobné, že tato skupina bude perspektivní. V menších studiích byl dokumentován antitrombotický účinek srovnatelný s dosud užívanou léčbou a s výjimkou razaxabanu, u něhož byl pro krvácivé komplikace zastaven vývoj, se ukazuje, že jde o léčbu bezpečnou s dobře predikovatelným účinkem.
Efekt nepřímých inhibitorů faktoru Xa spočívá v aktivaci antitrombinu III, přirozeného inhibitoru této proteázy. Jak bylo výše uvedeno, typickými představiteli aktivace antitrombinu III jsou hepariny. K potenciaci vazby antitrombinu III na FXa je nutná předchozí aktivace antitrombinu pentasacharidovou sekvencí molekuly heparinu. K této aktivaci je dostatečný 18sacharidový zbytek molekuly, ten pak reprezentuje selektivní inhibitor FXa. Léčiva této skupiny s osmnáctičlenným pentasacharidovým řetězcem označujeme jako pentasacharidy. Syntetický analog této sekvence je fondaparinux, další z nové generace, jež vyniká neobvykle dlouhým poločasem účinku, je idraparinux (dříve indaparinux). Fondaparinux je dobře dostupný po podkožní aplikaci, metabolizován je stejně jako hepariny, v ledvinách. Efekt je stabilní a nevyžaduje monitorování. Efekt byl prověřen v profylaxi trombózy hlubokého systému u ortopedických operací, v této indikaci je řadu let užíván. Ve srovnání s nízkomolekulárním heparinem enoxaparinem byl efekt na incidenci tromboembolických příhod stejný či lepší (REMBRANDT aj.). Nově je uvolněn a hrazen i pro ambulantní léčbu flebotrombózy.
Ve studii OASIS-6 bylo pozorováno snížení mortality a reinfarktu u nemocných s infarktem myokardu s elevací ST (STEMI) při léčbě fondaparinuxem, efekt byl výrazný u nemocných léčených konzervativně. Též u nemocných léčených koronární intervencí se ukázal příznivý trend snížení reokluze tepny ošetřené primární angioplastikou ve srovnání s heparinem (PENTALYSE). Recentně publikovaná Yusufova studie u 20 tisíc nemocných s nestabilními formami ICHS ukázala, že léčba fondaparinuxem byla stejně účinná na pokles koronárních příhod jako léčba enoxaparinem, výskyt závažného krvácení však byl poloviční. Lze tedy konstatovat, že léčba fondaparinuxem je účinná, tolerance byla dobrá a incidence krvácivých příhod je nižší než při standardní léčbě enoxaparinem. Závěrem je nutno uvést nevýhodu, která se týká nejen fondaparinuxu, ale i výše uvedených přímých inhibitorů FXa. Tou je absence neutralizačního antidota; efekt podání rekombinantního faktoru VII, který byl prověřován, nebyl zcela dostačující.
Mladším analogem fondaparinuxu je idraparinux. Modifikací molekuly pentasacharidu bylo dosaženo prodloužení poločasu až na 400 hod., a tak je schůdná možnost podkožního podávání pouze 1krát týdně. Významným pokrokem je skutečnost, že elegantním zákrokem, který není bohužel uvolněn k publikaci, bylo dosaženo možnosti rychlé neutralizace idraparinuxu antidotem. Možnost ukončení účinku při tak dlouhém působení je skutečně zásadního významu. Účinek je prověřován v indikaci profylaxe recidivy plicní embolie (Van Gogh-PE) a žilní trombózy (Van Gogh-DVT). Zda bude nebývale dlouhý efekt přínosem, ukáží výsledky srovnání s warfarinem. Studie, která sledovala efekt idraparinuxu v indikaci prevence tromboembolizační mozkové příhody u nemocných s fibrilací síní, byla bohužel předčasně ukončena, důvod ukončení mi není znám. Idraparinux je dalším slibným léčivem v řadě nových antikoagulancií.
Inhibitory tkáňového faktoru a ostatních faktorů koagulační kaskády
Tkáňový faktor (TF) není pouze iniciační molekulou v koagulační kaskádě, je též důležitým cytokinem účastnícím se zánětlivých pochodů. Koagulace iniciovaná TF má též svůj kontrolní systém brzdící jeho aktivitu. Tím je inhibitor tkáňového faktoru (tissue factor pathway inhibitor – TFPI). Tkáňový faktor hraje úlohu v řadě patologických procesů. Vedle destabilizace plátu a iniciace vlastní hemokoagulace se účastní i při jiných život ohrožujících procesech. Uvolnění TF u septických stavů spouští obávanou intravaskulární koagulaci. Vysoká aktivita TF je charakteristická pro nemocné s akutními koronárními syndromy a je spojena s horší prognózou. Nabízí se tedy inhibice tkáňového faktoru jako perspektivní léčebná metoda. K blokádě tkáňového faktoru byla navržena řada postupů, žádný však zatím nebyl schválen pro klinické užití a jen málo z nich překročilo fázi preklinickou. Rekombinantní TFPI (tifakogin) či antikoagulační proteiny z nematod se zkoušely u septických stavů a u akutních koronárních syndromů. Relativně velké studie bohužel neukázaly přínos podání tifakoginu při septických stavech. Efekt analog TFPI je testován u nestabilní anginy pectoris (ANTHEM/TIMI32), výsledky zatím nebyly zveřejněny. Alternativní postupy představuje podání protilátek proti TF či komplexu TF:FVIIa (NAPc2), ani zde však studie nepokročily do fáze časného klinického hodnocení. Postupy na bázi transferu genu pro TFPI se neosvědčily a nejsou zprávy, že by se v testování pokračovalo.
Pro úplnost přehledu je nutno uvést též inhibitory faktoru XIIIa, který katalyzuje vzájemnou vazbu vláken polymeru fibrinu, a ta se tak stávají rezistentnější k fibrinolýze plazminem. Podání inhibitorů tohoto faktoru tak činí trombus vnímavější k působení plazminu. Antikoagulancia tohoto typu jsou zatím ve stadiu preklinického zkoušení. Jak bylo řečeno, testován byl protein ze slin pijavice Haementeria ghilianii – tridegin – a několik menších syntetických molekul. Bohužel, poslední zprávy o výsledcích pocházejí z konce devadesátých let, je tedy pravděpodobné, že vývoj významně nepokročil.
Přípravky (drotekogin) s obsahem aktivovaného proteinu C, přirozeného faktoru stimulujícího degradaci koagulačních faktorů Va a VIIIa, jsou již užívány v léčbě kriticky nemocných v šokových septických stavech.
Závěr
Shrneme-li dosažený pokrok a perspektivy další léčby, pak je zřejmé, že v oblasti léčby dyslipidémií se v posledních letech mnoho nepokročilo a nezdá se, že by nás v nejbližší době čekala převratná, revolučně nová hypolipidemika. Světlou výjimkou byl ezetimib, zde však musíme ještě počkat na výsledky probíhajících prognostických studií. V léčbě dyslipidémií máme již nyní účinná léčiva, nicméně nepoužíváme je vždy racionálně, a zejména ne všem indikovaným nemocným jsou podávána.
V oblasti antitrombotik je situace lepší, rýsuje se nový thienopyridinový blokátor destičkových receptorů pro ADP prasugrel a non-thienopyridinový kangrelor. Naopak v oblasti další generace perorálně účinných blokátorů receptorů GP IIb/IIIa přešlapujeme na místě a o nové nadějné molekule nejsou zprávy. Výrazně lepší je situace v oblasti antikoagulancií, zejména ve skupině perorálně účinných antikoagulancií. Vyzdvihněme jen přímé blokátory FXa rivaroxaban a apixaban či přímý inhibitor trombinu dabigatran.
Ostatní oblasti kardiologie nebylo možno pro omezený rozsah uvést, ale i zde je řada léčiv před zavedením do klinické praxe a řada nadějných molekul se nachází ve fázi klinického hodnocení. Vzpomeňme jen nové antihypertenzivum z řady inhibitorů reninu – aliskiren, nové diuretikum ze skupiny inhibitorů vazopresinu (aquaretika) – tolvaptan – nebo blokátor endokanabinoidních receptorů – rimonaband. Těmto oblastem bude nutno věnovat samostatný přehled.