Působení léčiva na úrovni ovlivnění funkce enzymu
Článek věnovaný účinku léku na úrovni enzymu je druhým v řadě a navazuje na předchozí přehled účinků léčiv působících na receptor. Jsou probrány nejčastější typy reverzibilní inhibice enzymu (kompetitivní, nekompetitivní a akompetitivní) stejně jako inhibice nevratná. Na konkrétních příkladech blízkých klinikovi jsou ukázány přednosti jednotlivých typů působení a problémy s nimi spojené. Další části jsou věnovány léčivům navozujícím aktivaci enzymu, respektive přímé substituci enzymu. Poslední část se zabývá specifickými situacemi, které ovlivňují výsledný efekt, například lokalizaci cílového enzymu či význam transportních a metabolických systémů na výsledný účinek.
Léčiva působí velmi různorodým způsobem (tab. 1). V předchozím čísle časopisu Remedia byl shrnut mechanismus účinku léku působ ícího ovlivněním funkce receptoru. Tento přehled, který je určen především klinikům, volně navazuje na předchozí téma a věnuje se působení léčiva ovlivněním enzymu. Mechanismus účinku na úrovni ovlivnění enzymatické aktivity (či přímo substituce enzymu) představuje druhý nejčastější typ farmakodynamické aktivity.
ícího ovlivněním funkce receptoru. Tento přehled, který je určen především klinikům, volně navazuje na předchozí téma a věnuje se působení léčiva ovlivněním enzymu. Mechanismus účinku na úrovni ovlivnění enzymatické aktivity (či přímo substituce enzymu) představuje druhý nejčastější typ farmakodynamické aktivity.
Enzymy katalyzují řadu biologických pochodů, jako je trávení potravy, syntéza a degradace proteinů, glycidů, lipidů a dalších stavebních kamenů, inaktivují, degradují či aktivují léčiva či jiná xenobiotika, účastní se na transkripci RNA a na řadě dalších dějů. Jsou to zpravidla globulární proteiny, vzácněji glykoproteiny, o 60–2500 aminokyselinách. Enzymy snižují aktivační energii potřebnou k biologickým reakcím, které tak zásadně urychlují (obr. 1). Inhibice enzymu danou reakci zabrzdí, aktivace naopak urychlí, substituce vlastní děj umožní.
Podle zprostředkované reakce dělíme enzymy do několika skupin: oxidoreduktázy katalyzují oxidačně-redukční reakce, transferázy přenášejí funkční skupiny (například methyl-, acetyl- nebo fosfátovou skupinu), hydrolázy katalyzují hydrolýzu chemických vazeb, lyázy štěpí chemické vazby jiným způsobem než hydrolýzou, izomerázy katalyzují izomerizační reakce a ligázy spojují dvě molekuly kovalentní vazbou. Z přehledu je patrná široká plejáda možností, jak farmakologicky zasáhnout. Dle charakteru vztahu enzymu a léčiva rozlišujeme inhibitory a aktivátory enzymu. Některá léčiva pak mají vlastní enzymatickou aktivitu či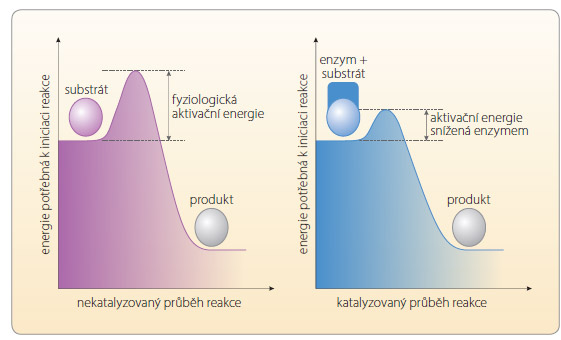 působí jako kofaktory ovlivňující funkci enzymu. Vedle typu reakce, kterou ovlivňujeme, a vedle charakteru působení (aktivace, inhibice) je důležitý též mechanismus zásahu (např. vratné či nevratné působení), kterým je určena intenzita a délka ovlivnění, respektive závislost účinku na koncentraci substrátu.
působí jako kofaktory ovlivňující funkci enzymu. Vedle typu reakce, kterou ovlivňujeme, a vedle charakteru působení (aktivace, inhibice) je důležitý též mechanismus zásahu (např. vratné či nevratné působení), kterým je určena intenzita a délka ovlivnění, respektive závislost účinku na koncentraci substrátu.
Inhibice enzymu
Při inhibici enzymu můžeme zasáhnout vratně či nevratně přímo působením na vlastní enzym. Tento mechanismus účinku je zcela běžný, příkladem jsou inhibitory angiotenzin konvertujícího enzymu (ACE), antikoagulancia, inhibitory cyklooxygenázy (COX) aj. Vedle přímého působení na enzym, respektive na jeho katalytické a vazebné místo, můžeme inhibovat či aktivovat expresi enzymu ovlivněním řídícího genu. Také s tímto typem ovlivnění se v praxi setkáváme často – řada léčiv může aktivovat či inhibovat izoenzym CYP3A4 působením na pregnanový receptor, který řídí transkripci jeho genu. Tomuto typu inhibice bude věnováno místo v samostatném přehledu o transportu a transformaci
léčiva.
Inhibice vlastního enzymu může mít různý charakter daný typem vazby (reverzibilní či ireverzibilní) a místem vazby inhibitoru na enzym (na katalytické centrum, v blízkosti centra či vazba až na komplex enzym/substrát).
Reverzibilní inhibitory
Reverzibilní inhibitory jsou volně disociovatelné z vazby, aktivita enzymu je ovlivněna jen dočasně, vazba je zpravidla nekovalentní – elektromagnetická či hydrofobní. Podle vztahu inhibitor-enzym-substrát rozlišujeme několik typů působení. Zásadní rozdíly mezi nimi jsou dány vlivem koncentrace substrátu na účinek inhibitoru: zvýšení koncentrace substrátu sníží účinek, neovlivní účinek, či zvýší účinek inhibitoru. V biologických systémech i ve farmakologii se setkáváme se všemi typy.
Kompetitivní inhibice
Kompetitivní inhibice je charakterizována vzájemným soutěžením inhibitoru a substrátu o katalytické místo enzymu (obr. 2). Efekt inhibice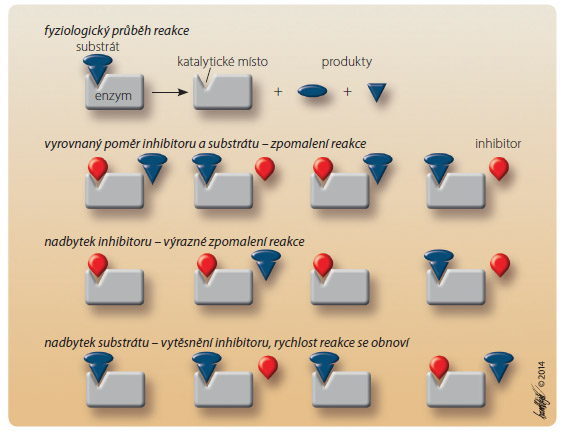 závisí na poměru substrátu k inhibitoru, tedy vysoká koncentrace substrátu vytěsní inhibitor z vazby a naopak. Inhibice enzymu vede zpravidla ke zvýšení koncentrace substrátu a ke snížení výsledného účinku léčby.
závisí na poměru substrátu k inhibitoru, tedy vysoká koncentrace substrátu vytěsní inhibitor z vazby a naopak. Inhibice enzymu vede zpravidla ke zvýšení koncentrace substrátu a ke snížení výsledného účinku léčby.
V případě kompetice je struktura inhibitoru obvykle podobná substrátu. Příkladem kompetitivní inhibice je blokáda enzymu hydroxymethyl-glutaryl-CoA-reduktázy (HMG-CoA-reduktáza), tzv. rate-limiting kroku syntézy cholesterolu. Podobnost substrátu – HMG-CoA – a farmakoforu statinů je vysoce nápadná (obr. 3). Pro jejich efekt je rozhodující koncentrace v místě syntézy cholesterolu – na úrovni ribozomů jaterních buněk. Vedle stupně inhibice aktivity reduktázy je při kompetitivní inhibici důležitá též délka působení, tedy zajištění účinku během celého dávkovacího intervalu. Je-li inhibice nedostatečná a obnoví-li se aktivita reduktázy, dojde k rychlé resyntéze cholesterolu a hypolipidemický efekt se snižuje. To vysvětluje např. rozdíly v efektu rosuvastatinu a atorvastatinu, statinů s dlouhou dobou účinku (t1/2 kolem 20 hod.), a simvastatinu, lovastatinu či fluvastatinu s účinkem krátkodobým (t1/2 kolem 2 hod.).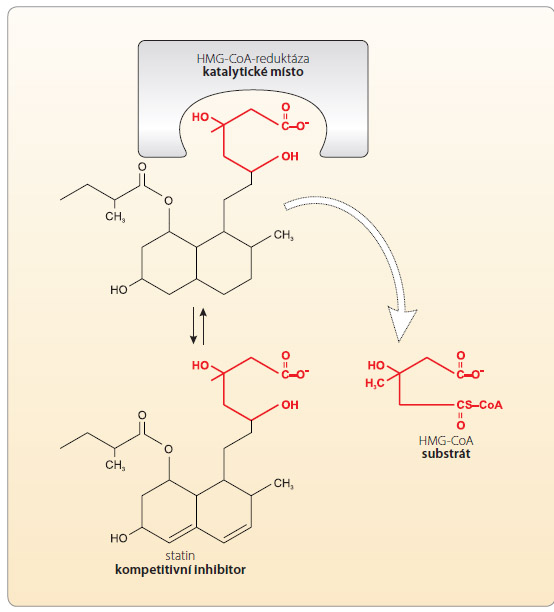
Nekompetitivní inhibice
Nekompetitivní inhibice je dána vazbou inhibitoru na enzym v blízkosti katalytického místa. Navázáním na tzv. alosterické místo dojde ke změně prostorového uspořádání molekuly enzymu s inhibicí účinku (obr. 4). Efekt v tomto případě není závislý na poměru substrátu k inhibitoru, ale pouze na poměru inhibitoru a enzymu. Nekompetitivní inhibicí je charakterizován účinek některých léčiv (např. antibiotik, diuretik a antidepresiv) či jedů (např. organofosfátů a těžkých kovů).
Příkladem nekompetiv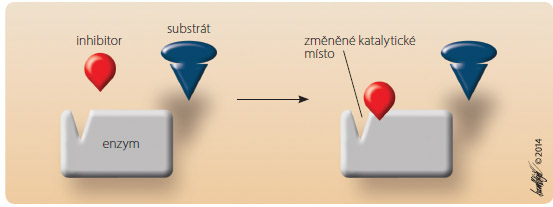 ní inhibice v účinku antibiotik jsou peptidické inhibitory aminoglykosidázy. V působení antidepresiv jsou příkladem selektivní reverzibilní inhibitory monoaminooxidázy (IMAO) – RIMA (např. moklobemid), naproti tomu klasické IMAO inhibují vlastní enzym ireverzibilní kovalentní vazbou. Furosemid je zajímavým příkladem diuretika, které působí v závislosti na pH, na koncentraci i na typu cílové pumpy jak kompetitivní, tak nekompetitivní inhibicí. Hlavní efekt je dán kompetitivní inhibicí zpětného vstřebávání natria a kalia blokádou Na-K-2Cl symportéru, naopak ovlivnění chloridového transportu blokádou Cl–/HCO3– -ATPázy či blokáda GABA receptorů v CNS má charakter nekompetitivní inhibice.Nekompetitivní inhibice je významná též při lékových interakcích. Například na úrovni inhibice oxidázy CYP2C9 působí tímto způsobem flavonoidy či nifedipin.
ní inhibice v účinku antibiotik jsou peptidické inhibitory aminoglykosidázy. V působení antidepresiv jsou příkladem selektivní reverzibilní inhibitory monoaminooxidázy (IMAO) – RIMA (např. moklobemid), naproti tomu klasické IMAO inhibují vlastní enzym ireverzibilní kovalentní vazbou. Furosemid je zajímavým příkladem diuretika, které působí v závislosti na pH, na koncentraci i na typu cílové pumpy jak kompetitivní, tak nekompetitivní inhibicí. Hlavní efekt je dán kompetitivní inhibicí zpětného vstřebávání natria a kalia blokádou Na-K-2Cl symportéru, naopak ovlivnění chloridového transportu blokádou Cl–/HCO3– -ATPázy či blokáda GABA receptorů v CNS má charakter nekompetitivní inhibice.Nekompetitivní inhibice je významná též při lékových interakcích. Například na úrovni inhibice oxidázy CYP2C9 působí tímto způsobem flavonoidy či nifedipin.
Akompetitivní inhibice
Akompetivní inhibice je podobná nekompetitivní inhibici, inhibitor se však váže až na komplex enzymu a substrátu, stejně však dojde ke změně alosterického uspořádání molekuly enzymu a k inhibici přeměny substrátu na produkt (obr. 5). Zatímco při nekompetitivní inhibici se mohl i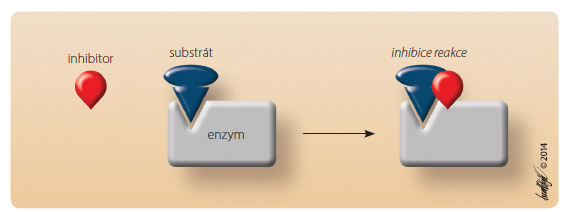 nhibitor vázat na enzym i při nepřítomnosti substrátu, v případě akompetitivní inhibice je vazba umožněna až při určité koncentraci substrátu. Efekt této inhibice stoupá při vyšší koncentraci substrátu. Tento typ inhibice je velmi vzácný, příkladem je působení lithia na metabolismus inositolu, který je dán akompetitivní inhibicí inositol monofosfatázy.
nhibitor vázat na enzym i při nepřítomnosti substrátu, v případě akompetitivní inhibice je vazba umožněna až při určité koncentraci substrátu. Efekt této inhibice stoupá při vyšší koncentraci substrátu. Tento typ inhibice je velmi vzácný, příkladem je působení lithia na metabolismus inositolu, který je dán akompetitivní inhibicí inositol monofosfatázy.
Ireverzibilní inhibitory
Ireverzibilní inhibitory se váží na cílový enzym kovalentní, eventuálně nekovalentní vazbou, ze které pouze velmi pomalu disociují. V molekule enzymu jsou to zejména serinové či histidinové části, na které se vlastní inhibitor váže a je tak prakticky nevratně vázán k oblasti katalytického místa enzymu. Aktivita enzymu je tlumena dlouhodobě a metabolická cesta je obnovena zpravidla až syntézou enzymu nového. Délka působení je tak dána schopností tkáně resyntetizovat nový enzym.
Příkladem nevratné inhibice je působení kyseliny acetylsalicylové (ASA) na cyklooxygenázu 1 (COX-1). Mechanismem inhibice je acetylace serinu v místě katalytického místa COX-1. Aktivita enzymu COX-1 je vysoká zejména v endotelu – kde je tvořen prostacyklin (PGI2), a v trombocytech – s výsledným produktem tromboxanem A2 (TXA2). Endotelie jako jaderná buňka rychle syntetizuje nový enzym COX-1 a syntéza prostacyklinu se rychle obnoví. Naopak trombocyt jako bezjaderný element již není schopen syntézy nového enzymu a destička není schopna po dobu další cirkulace v oběhu tvořit nový tromboxan A2. V důsledku toho je agregace defektní po celou dobu cirkulace trombocytu. Poměr PGI2/TXA2 tak po aplikaci ASA stoupá a převáží vazodilatační a protidestičkový účinek. Aby situace nebyla tak jednoduchá, je ASA schopna acetylovat i jiné enzymy či transkripční faktory. Navíc aktivní metabolit vzniklý po deacetylaci ASA „aspirinovými“ esterázami v enterocytu a hepatocytu – kyselina salicylová – působí inhibici COX-1 potlačením exprese enzymu na úrovni řídícího genu [1]. Mateřská molekula tak působí ireverzibilní inhibici COX-1 a aktivní metabolit tlumí expresi řídícího genu.Nevratná inhibice enzymu je mechanismem účinku také řady antibiotik. Např. penicilin se kovalentně váže na bakteriální transpeptidázu (enzym nutný k vzájemné vazbě peptidů typu cross-linking) a tak zabrání syntéze akteriální stěny.
Aktivace enzymu
Aktivace enzymu je častým mechanismem řízení biologických dějů, s nímž se setkáme i při působení léčiv. Zpravidla je aktivace enzymu zprostředkována specifickým receptorem, nicméně volně pronikající gasotransmitery, jako je oxid dusnatý (NO), oxid uhelnatý (CO) a další, mohou působit přímo na enzym. V praxi užíváme nitráty (nitroglycerin, isosorbid dinitrá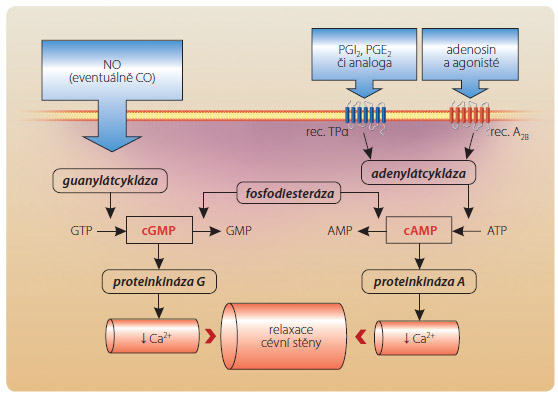 t aj.) a donátory NO (molsidomin, nebivolol aj.), které zvyšují nabídku NO. Oxid dusnatý působí přímo aktivaci guanylátcyklázy, zvýší nabídku cyklického guanosinmonofosfátu (cGMP) a navodí azodilataci, bronchodilataci, sníží kontraktilitu myokardu, inhibuje aktivaci trombocytů či spustí plejádu dalších pochodů (obr. 6). Podobně, při nutnosti reaktivace acetylcholinesterázy (po otravě organofosfáty či při předávkování inhibitory acetylcholinesterázy), užíváme pralidoxim, který navozením konformačních změn obnoví funkci enzymu.Aktivace enzymu může probíhat přímo, či nepřímo – teprve v komplexu s biologickým regulátorem. Příkladem je mechanismus funkce heparinu a jeho derivátů – ty se váží na antitrombin a teprve vzniklý komplex inhibuje dvě významné proteázy účastnící se hemostázy – trombin či aktivovaný faktor X.
t aj.) a donátory NO (molsidomin, nebivolol aj.), které zvyšují nabídku NO. Oxid dusnatý působí přímo aktivaci guanylátcyklázy, zvýší nabídku cyklického guanosinmonofosfátu (cGMP) a navodí azodilataci, bronchodilataci, sníží kontraktilitu myokardu, inhibuje aktivaci trombocytů či spustí plejádu dalších pochodů (obr. 6). Podobně, při nutnosti reaktivace acetylcholinesterázy (po otravě organofosfáty či při předávkování inhibitory acetylcholinesterázy), užíváme pralidoxim, který navozením konformačních změn obnoví funkci enzymu.Aktivace enzymu může probíhat přímo, či nepřímo – teprve v komplexu s biologickým regulátorem. Příkladem je mechanismus funkce heparinu a jeho derivátů – ty se váží na antitrombin a teprve vzniklý komplex inhibuje dvě významné proteázy účastnící se hemostázy – trombin či aktivovaný faktor X.
Přímé působení léčiva jako enzymu
Řada léčiv má charakter enzymu – jak přirozeného, tak jeho analoga. Prvou skupinou jsou léčiva podávaná za účelem navození pochodu stimulovaného aktivací daného enzymu. Zde jsou příkladem fibrinolytika, serinové proteázy aktivující jinou proteázu – plazminogen – na plazmin (obr. 7). Plazmin má cílovou fibrinolytickou aktivitou: štěpí fibrin na degradační produkty (např. D-dimery). Užíváme buď přirozený aktivátor tkáňového p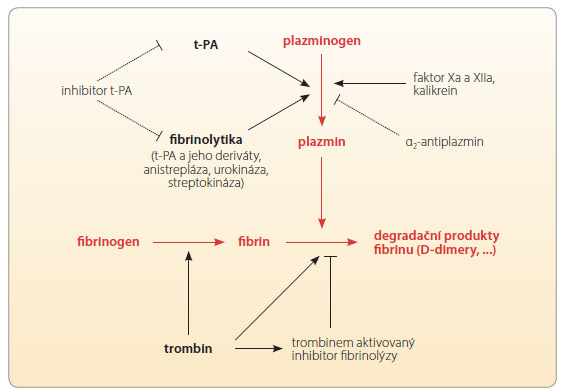 lazminogenu (tPA) alteplázu, jeho analoga (např. reteplázu či tenecteplázu) nebo aktivátory jiného, např. bakteriálního původu (streptokinázu). Preference působení v místě vlastního trombu či systémový efekt rozhoduje o případných nežádoucích účincích. Jak již bylo ukázáno, mnohé proteázy nejsou selektivní pro daný protein, ale štěpí peptidickou vazbu mezi konkrétními aminokyselinami v určitém pořadí peptidového řetězce. Peptidy, které splňují tuto podmínku, jsou pak substráty této peptidázy. Působení na plejádu cílových peptidů je charakteristické i pro fibrinolytika a ovlivňuje výsledný účinek. Pro působení fibrinolytik – aktivátorů plazminu – je významná lokalizace jejich působení na cílovou molekulu, tedy poměr systémového účinku a účinku v místě trombu. Ten ovlivňuje míru účinku na ostatní systémy než jen na hemostázu.
lazminogenu (tPA) alteplázu, jeho analoga (např. reteplázu či tenecteplázu) nebo aktivátory jiného, např. bakteriálního původu (streptokinázu). Preference působení v místě vlastního trombu či systémový efekt rozhoduje o případných nežádoucích účincích. Jak již bylo ukázáno, mnohé proteázy nejsou selektivní pro daný protein, ale štěpí peptidickou vazbu mezi konkrétními aminokyselinami v určitém pořadí peptidového řetězce. Peptidy, které splňují tuto podmínku, jsou pak substráty této peptidázy. Působení na plejádu cílových peptidů je charakteristické i pro fibrinolytika a ovlivňuje výsledný účinek. Pro působení fibrinolytik – aktivátorů plazminu – je významná lokalizace jejich působení na cílovou molekulu, tedy poměr systémového účinku a účinku v místě trombu. Ten ovlivňuje míru účinku na ostatní systémy než jen na hemostázu.
Plazmin je uvolněn v játrech do systémového oběhu jako proenzym (zymogen) – plazminogen ve dvou izoformách (I a II). Ty se liší počtem glykosylovaných míst a afinitou k rozdílným strukturám. Typ I má větší afinitu k povrchu trombu, zatímco typ II se váže méně specificky na buněčné membrány endotelu apod. Právě tato vazba na povrch změní terciární strukturu plazminogenu a umožní jeho aktivaci na plazmin. I když je působení různých fibrinolytik stejné, všechna štěpí peptidickou vazbu mezi argininem a valinem (Arg-561 a Val-562), výsledný farmakodynamický efekt se liší. Afinita první generace fibrinolytik (zejména streptokinázy) má výrazně menší selektivitu k I. typu plazminogenu, a generovaný plazmin má proto daleko větší systémový účinek. Díky tomu tato proteáza aktivuje či degraduje řadu působků s vazoaktivním, hemostatickým či prozánětlivým účinkem. Aplikace streptokinázy je tak spojena s vyšším výskytem nežádoucích účinků, zejména s hypotenzí, než podávání druhé generace fibrinolytik, která mají naopak vysokou afinitu k plazminogenu vázanému na povrch trombu, a účinek je místně omezen.
Další velkou skupinou léčiv působících na úrovni enzymu je substituce defektních či zcela chybějících enzymů. Například u různých typů hemofilie aplikujeme příslušný koagulační faktor, u lyzozomálních chorob substituujeme chybějící enzym, např. α-galaktosidázu či glukocerebrosidázu, nebo při poruše zevní sekrece pankreatu podáváme pankrea-
tické enzymy.
Význam lokalizace cílového enzymu
Vlastní katalytická reakce může probíhat v různých prostředích – v krvi, v tkáňové tekutině, v mozkomíšním moku, intracelulárně, intranukleárně či mimo tkáně, např. v trávicím traktu. Pro výsledný farmakologický efekt je rozhodující nabídka inhibitoru, aktivátoru či aplikovaného enzymu v místě působení. Je-li enzym lokalizován na membránách endoteliálních buněk cév či je přímo v krvi (v samotné plazmě či např. na membráně trombocytů) a katalytická reakce probíhá extracelulárně, je významná koncentrace léčiva v plazmě či v tkáňovém moku. O té rozhoduje dávka léčiva, biologická dostupnost, rychlost degradace a eliminace, afinita léčiva k enzymu, délka vazby léčiva na enzym, nabídka enzymu a substrátu apod. Příkladem léčiva působícího na úrovni membrány endoteliálních buněk jsou inhibitory ACE. Příkladem léčiva působícího v plazmě jsou přímé i nepřímé inhibitory trombinu či faktoru Xa (heparin, xabany, gatrany aj.), koagulační faktory a plejáda dalších látek.
Je-li však enzym lokalizován intracelulárně, je rozhodující koncentrace v místě působení – v jaterních mikrozomech, na membráně mitochondrií svalových buněk, v lyzozomech, v mozkomíšním moku a podobně. Zajímavým příkladem působení na úrovni intracelulárně umístěného enzymu jsou statiny. Klíčový enzym, HMG-CoA-reduktáza, je lokalizován zejména v mikrozomech hepatocytu. O terapeutickém účinku jejich inhibitorů pak rozhoduje celá řada faktorů. Na úrovni dostupnosti statinů hraje úlohu aktivita efluxních pump a metabolických systémů v enterocytu (zejm. P-gp, CYP3A4 a CYP2C9), vstup do hepatocytu je facilitován transportérem OATP 1B1 (organic aniont transporting polypeptide 1B1) a řadou dalších influxních pump. Aktivita vlastního cílového enzymu HMG-CoA-reduktázy a její citlivost k inhibici je ovlivněna řadou polymorfismů vlastního enzymu i aktivitou genů řídících její expresi. Délka působení je ovlivněna aktivitou eliminačních pump na žlučovém pólu hepatocytu, které vylučují statiny do žluče (zejména P-gp), eventuálně v nefronu do moče, a v neposlední řadě jsou statiny inaktivovány oxidázami systému CYP či transferázami v játrech (obr. 8). Dále se uplatň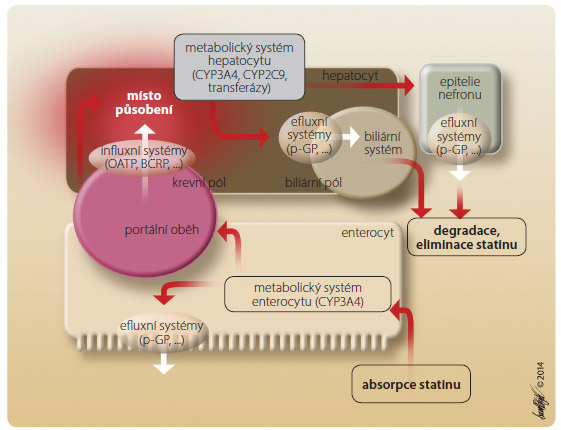 uje celá plejáda faktorů určujících absorpci cholesterolu, jeho syntézu, transport do tkání (včetně transportu reverzního) či jeho degradaci. Tyto vlivy jsou dnes předmětem intenzivního výzkumu.
uje celá plejáda faktorů určujících absorpci cholesterolu, jeho syntézu, transport do tkání (včetně transportu reverzního) či jeho degradaci. Tyto vlivy jsou dnes předmětem intenzivního výzkumu.
Relativně více je známo o vlivu poruch facilitovaného hepatálního transportu na účinnost a bezpečnost statinů. Pro hypolipidemický účinek statinů je rozhodující koncentrace na ribozomech jaterních buněk, tj. v místě syntézy cholesterolu. Na úrovni hepatocytu je transmembranózní přesun statinů potencován influxním přenašečem OATP 1B1. Na krevním pólu hepatocytu tento protein umožní vstup statinu do hepatocytu, na biliárním pólu pak další pumpy – zejména efluxní transportér glykoprotein P (P-gp) – umožní eliminaci do žluče.
Afinita obou pump k různým statinům je odlišná. Nejvíce je stimulován vstup simvastatinu, nejméně fluvastatinu a rosuvastatinu (simva- > pitava- > atorva- > prava- > rosuva- > fluvastatin) [2, 3]. Nízká aktivita transportéru OATP 1B1 na základě farmakogenetické variability či při jeho inhibici (např. některými antibiotiky, sartany či perorálními antidiabetiky) vede k nedostatečné koncentraci statinu v místě působení a ke sníženému účinku [4]. Klinicky významná je tato skutečnost u statinu s vysokou afinitou k transportérům – simvastatinu. Na druhé straně je transport do hepatocytu podmínkou eliminace statinu do žluče se zapojením zejména P-gp. Vysoká hladina simvastatinu u nositelů varianty řídícího genu SLCO1B1 (rs4149056) se sníženou aktivitou pumpy je spojena s vysokým výskytem myalgií a myopatií. V heterozygotní formě (alely TC) se tento polymorfismus vyskytuje asi u 15 % indoevropské populace a v homozygotní formě s nulovou aktivitou (alely CC) asi u 2 % populace. Zvýšení frekvence výskytu myalgií/myopatií pak bylo u těchto variant typu „loss-of-function” při léčbě simvastatinem pětinásobné, respektive dvacetinásobné.
Jiným příkladem významu cílového místa působení enzymu je substituční enzymatická léčba. V případě substituce koagulačního faktoru (např. při hemofilii) je situace jednoduchá, stačí dosáhnout minimální účinné koncentrace v krvi a hemostáza je obnovena. Daleko komplikovanější je stav u substituce enzymů při již zmíněných lyzozomálních chorobách. Zde je potřeba dosáhnout účinné koncentrace enzymu v místě intracelulárního působení, tj. v lyzozomu. Jinými slovy, je potřeba aktivovat endocytózu enzymu do buňky jejich vazbou na transportní polypeptidy – endocytární receptory (např. k manózo-6-fosfátovému či k sortilinovému receptoru). Afinita substituovaného enzymu k těmto přenašečům a kapacita přenašeče pak ovlivňuje vlastní klinický efekt. Například u Fabryho choroby je přítomen deficit lyzozomálního enzymu α-galaktosidázy. Ta se skládá z proteinu (zodpovědného za katalytickou funkci) a postranních oligosacharidových řetězců (ovlivňujících afinitu k pumpám, tedy farmakokinetické vlastnosti). K substituci jsou užívány enzymy vzniklé rekombinantní technologií od různých výrobců. Ač je proteinová část shodná, rozdíly jsou v postranních oligosacharidových řetězcích. Tak je dána rozdílná afinita k endocytárním přenašečům určujícím lyzozomální koncentraci enzymu či jeho eliminaci.
Závěr
Roztřídíme-li působení léčiv do logických celků, zdá se, že je situace přehledná a účinek se dá spolehlivě určit. Bohužel je situace – i z pohledu působení léku na úrovni vlastního enzymu – daleko komplikovanější. V dalším volně navazujícím článku v následujícím čísle časopisu Remedia se podíváme blíže na problematiku inhibitorů angiotenzin konvertujícího enzymu, respektive na problematiku inhibice systému renin-angiotenzin-aldosteron (RAA). Tento model byl vybrán nejen proto, že se v případě inhibitorů ACE jedná o léčivo působící na úrovni enzymu a v případě sartanů o léčivo působící na úrovni receptoru. Srovnání uvnitř skupin a mezi skupinami je – díky velké řadě novinek v této oblasti – velmi zajímavé. Navíc jde o jednu z nejvýznamnějších skupin léků v klinické medicíně.
Seznam použité literatury
- [1] Higgs GA, Salmon JA, Henderson B, et al. Pharmacokinetics of aspirin and salicylate in relation to inhibition of arachidonate cyclooxygenase and antiinflammatory activity. Proc Natl Acad Sci 1987; 84: 1417–1420.
- [2] Niemi M, Pasanen MK, Neuvonen PJ. Organic anion transporting polypeptide 1B1: a genetically polymorphic transporter of major importance for hepatic drug uptake. Pharmacol Rev 2011; 63: 157–181.
- [3] Wilke RA, Ramsey LB, Johnson SG, et al., and Clinical Pharmacogenomics Implementation Consortium (CPIC). The clinical pharmacogenomics implementation consortium: CPIC guideline for SLCO1B1 and simvastatin-induced myopathy. Clin Pharmacol Ther 2012; 92: 112–117.
- [4] Klatt S, Fromm MF, König J. The influence of oral antidiabetic drugs on cellular drug uptake mediated by hepatic OATP family members. Basic Clin Pharmacol Toxicol 2013; 112: 244–250.