Působení léčiva na úrovni receptoru
Cestou ovlivnění receptorů (aktivací či inhibicí) působí téměř polovina léčiv, která podáváme. Článek dává přehled o typech cílových receptorů, o jejich přímém ovlivnění či působení o etáž níže, na úrovni „druhého posla”, o významu interakce léčivo–receptor, interakce organismus–receptor či interakce organismus–léčivo (tj. uvedeny jsou důležité ukazatele farmakokinetické). V neposlední řadě jsou probrány farmakogenetické vlivy, které výsledný efekt léčby zásadně modifikují. Specifikou článku je výrazná orientace na klinický význam jednotlivých probíraných skutečností.
Z farmakologického pohledu jsou léky definovány jako chemické substance, které užíváme k léčbě, k profylaxi či z diagnostických důvodů. Očekáváme od nich zlepšení stavu ve smyslu kvality života či prognózy. Léčiva působí různými mechanismy, mohou ovlivnit regulační receptory, metabolické systémy (zejména enzymy, eventuálně kofaktory), iontové či transportní kanály, regulační proteiny apod. Cílovou molekulou jsou zpravidla proteiny – jako součást receptoru, iontového kanálu či přímo enzymu. V tomto stručném přehledu, orientovaném zejména na klinika, se budeme věnovat působení léčiva na úrovni receptoru, v dalším pokračování pak ovlivnění enzymů a iontových kanálů.
Reakce zprostředkované působením léčiva na receptor
Cestou aktivace či inhibice receptoru působí asi 40 % užívaných léčiv. Receptor, zpravidla umístěný na membráně (cytoplazmy buňky či jádra), eventuálně lokalizovaný volně v cytoplazmě, je protein schopný rozpoznat specifickou molekulu – ligand – a odpovědět na ni signálem, tedy buněčnou odpovědí. Vlastní odpověď může být přímá nebo zprostředkovaná další molekulou – druhým poslem.
Zatímco mezi enzymy či regulační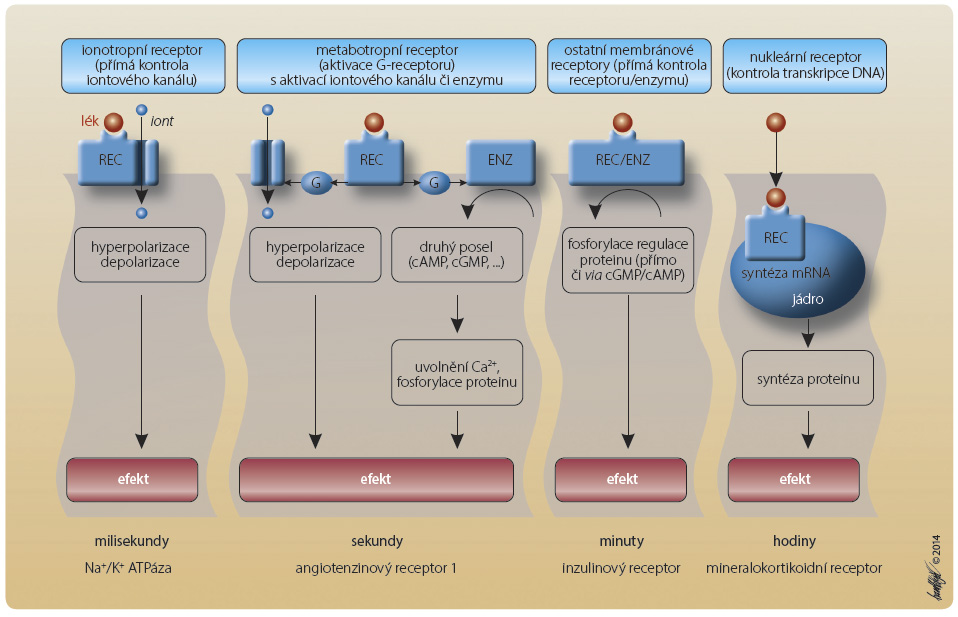 mi proteiny nejsou významnější rozdíly, struktura regulačních receptorů je velmi komplikovaná. Podle požadavků na rychlost reakce jsou membrány osazeny konkrétním typem ze základních čtyř variant. Prvou jsou receptory osazené na iontový kanál – ionotropní, druhou jsou receptory spřažené s G-proteinem či s enzymem – tzv. metabotropní, třetí jsou přímo či nepřímo působící membránové receptory a čtvrtou nukleární receptory kontrolující přímo přepis DNA na úrovni jádra (obr. 1). Odpověď na aktivaci receptoru je různě dlouhá, trvá od milisekund po hodiny a je závislá na typu receptoru.
mi proteiny nejsou významnější rozdíly, struktura regulačních receptorů je velmi komplikovaná. Podle požadavků na rychlost reakce jsou membrány osazeny konkrétním typem ze základních čtyř variant. Prvou jsou receptory osazené na iontový kanál – ionotropní, druhou jsou receptory spřažené s G-proteinem či s enzymem – tzv. metabotropní, třetí jsou přímo či nepřímo působící membránové receptory a čtvrtou nukleární receptory kontrolující přímo přepis DNA na úrovni jádra (obr. 1). Odpověď na aktivaci receptoru je různě dlouhá, trvá od milisekund po hodiny a je závislá na typu receptoru.
Mluvíme-li o receptorech, je nutno uvést důležitou skutečnost – odpověď receptorů (stejně jako enzymů) může být přímá – například depolarizace membrány – či zprostředkovaná „druhým poslem“. Systém druhých poslů je dán potřebou zajištění rychlé odpovědi pomocí malé, dobře difundující molekuly v situaci, kdy primární ligand není schopen aktivaci cílové struktury zajistit. Systém je postaven na regulačních proteinech (např. G-protein) či katalytických proteinech (např. adenylátcykláza či fosfolipáza C), které generují vlastní zprostředkovatelskou molekulu. Tou jsou např. ionty kalcia, oxid dusnatý (NO), cyklický adenosinmonofosfát (cAMP) či guanosinmonofosfát (cGMP). Receptory působící cestou druhých poslů můžeme ovlivňovat na různých etážích. Obdobného výsledku dosáhneme stimulací vlastního receptoru, působením na katalytický protein (enzym) ovlivňující syntézu či uvolnění druhého posla nebo inhibicí degradace molekuly druhého posla. Příkladem jsou protidestičkové léky. Z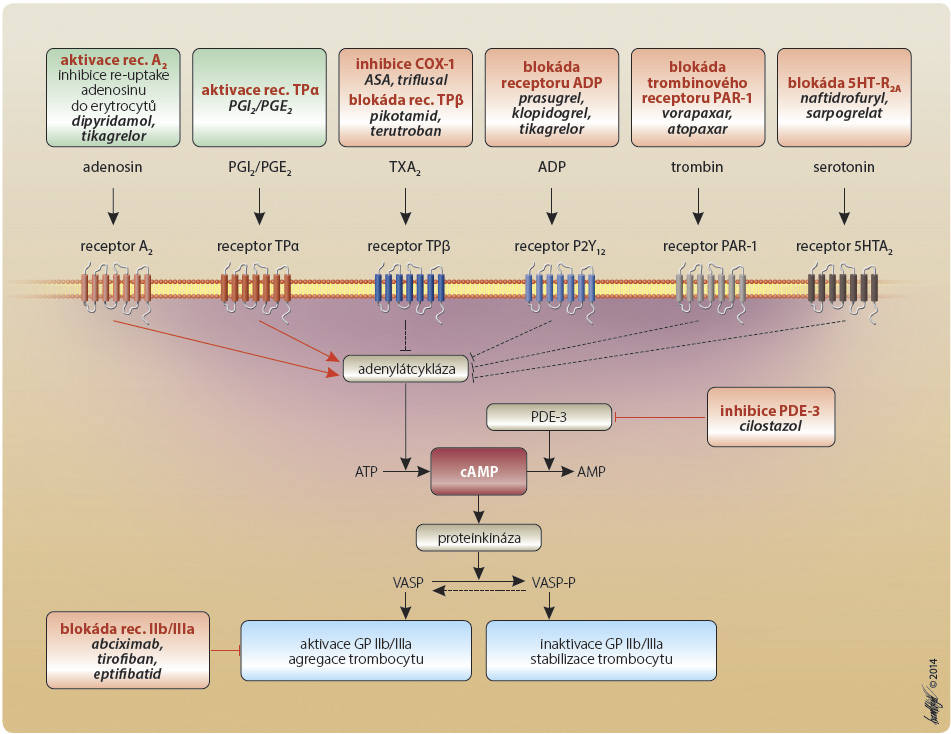 de potřebujeme zvýšit nabídku klíčové látky cAMP (obr. 2). Ta je syntetizována adenylátcyklázou (AC) a degradována fosfodiesterázou 3 (PDE-3). Adenylátcyklázu aktivujeme stimulací tromboxan/prostanoidového receptoru α (TPRα) nebo adenosinového receptoru A2R. Zvýšení nabídky AC vede k syntéze cAMP a ke stabilizaci trombocytu. Naopak AC je inhibována působením tromboxan/prostanoidového receptoru β (TPRβ), stimulací ADP receptoru P2Y12 či trombinového receptoru PAI-1. Poklesem nabídky cAMP je tromboxan aktivován a dochází k expresi vazných receptorů IIb/IIIa. Terapeuticky obdobný efekt tak má stimulace aktivačních receptorů (prostacyklinem či adenosinem) či blokáda receptorů inhibičních (klopidogrelem, prasugrelem, tikagrelorem, vorapaxarem apod.). Jak bylo řečeno, cAMP je v trombocytu degradován PDE-3. Tento izoenzym je účinně inhibován např. cilostazolem, lékem s antiagregačním a vazodilatačním účinkem. Uvedený mechanismus představuje druhou cestu k dosažení inhibice primární hemostázy. Konečně poslední cestou je přímé působení na cílovou strukturu, v tomto případě na vazné receptory IIb/IIIa, které pomocí bivalentního vazného proteinu umožní vlastní agregaci destiček. Jak je patrné, cesty k dosažení stejného výsledku mohou být různé.
de potřebujeme zvýšit nabídku klíčové látky cAMP (obr. 2). Ta je syntetizována adenylátcyklázou (AC) a degradována fosfodiesterázou 3 (PDE-3). Adenylátcyklázu aktivujeme stimulací tromboxan/prostanoidového receptoru α (TPRα) nebo adenosinového receptoru A2R. Zvýšení nabídky AC vede k syntéze cAMP a ke stabilizaci trombocytu. Naopak AC je inhibována působením tromboxan/prostanoidového receptoru β (TPRβ), stimulací ADP receptoru P2Y12 či trombinového receptoru PAI-1. Poklesem nabídky cAMP je tromboxan aktivován a dochází k expresi vazných receptorů IIb/IIIa. Terapeuticky obdobný efekt tak má stimulace aktivačních receptorů (prostacyklinem či adenosinem) či blokáda receptorů inhibičních (klopidogrelem, prasugrelem, tikagrelorem, vorapaxarem apod.). Jak bylo řečeno, cAMP je v trombocytu degradován PDE-3. Tento izoenzym je účinně inhibován např. cilostazolem, lékem s antiagregačním a vazodilatačním účinkem. Uvedený mechanismus představuje druhou cestu k dosažení inhibice primární hemostázy. Konečně poslední cestou je přímé působení na cílovou strukturu, v tomto případě na vazné receptory IIb/IIIa, které pomocí bivalentního vazného proteinu umožní vlastní agregaci destiček. Jak je patrné, cesty k dosažení stejného výsledku mohou být různé.
Ovlivnění terapeutické odpovědi na úrovni interakce receptor–léčivo
Při působení léčiva na receptor (ale obdobně i interakce s enzymem či iontovým kanálem) je důležitá jak afinita k receptoru, tak povaha vazby na receptor (resp. obecně na cílovou strukturu). Afinita je dána silou vazby na receptor – při vysoké afinitě obsadí ligand při stejné koncentraci větší počet receptorů než při afinitě nízké. Při vyšší afinitě též setrvává ligand déle na vlastním receptoru, tedy má delší disociační poločas.
Příkladem významu afinity k receptoru může být blokátor trombocytárních receptorů IIb/IIIa abciximab. Jeho vazba na receptor je tak silná, že přetrvává prakticky po dobu cirkulace trombocytu. Tak i při plazmatickém poločasu kolem 10 minut trvá protidestičkový efekt několik dnů. Jindy, při afinitě nižší, je naopak výsledný účinek léčiva dán aktuální koncentrací v místě působení. Například u blokátorů angiotenzinových receptorů AT1 (sartanů) jsou rozdíly v setrvání ve vazbě na receptor málo významné, například disociační poločas vazby se pohybuje u všech sartanů v rozmezí 100–200 minut, kdežto rozdíly v plazmatickém poločase jsou větší – v rozpětí 4–24 hodin. Efekt telmisartanu trvá řádově 48 hodin a při případném jednorázovém vynechání dávky léku účinek neodezní, naopak u enalaprilu či valsartanu se již po 12 hodinách efekt snižuje, a proto je výhodnější aplikace ve dvou denních dávkách.
Vazba na receptor může být vratná či nevratná. Pokud je vazba ireverzibilní, je receptor trvale vyřazen z funkce. Výsledný farmakodynamický efekt, respektive jeho délka, potom závisí na schopnosti a rychlosti tkáně syntetizovat jiný receptor. Velká většina tkání je schopna restituce funkce, nicméně např. bezjaderný trombocyt postrádá DNA k přepisu informace nutné pro syntézu nového receptoru (či obdobně enzymu) a trombocyt zůstává po dobu své cirkulace afunkční. Příkladem je např. blokáda trombocytárního receptoru P2Y12 pro adenosindifosfát (ADP). Ta může být ireverzibilní (po podání klopidogrelu či prasugrelu) i reverzibilní (po tikagreloru). Výhoda dlouhodobé inhibice destičkových funkcí vyvstane u zapomětlivého nemocného, kdy vynechání dávky nebude mít zásadní dopad na aktivaci primární hemostázy, naopak krátkodobější reverzibilní blokádu oceníme při krvácení, kdy antiagregační efekt odezní do 24 hodin. U většiny léků je cílová struktura (receptor, případně kanál, enzym) ovlivněna reverzibilně.
Vlas
{kind=link}
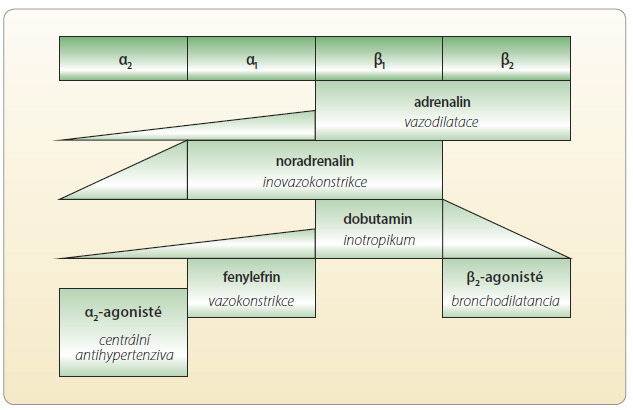
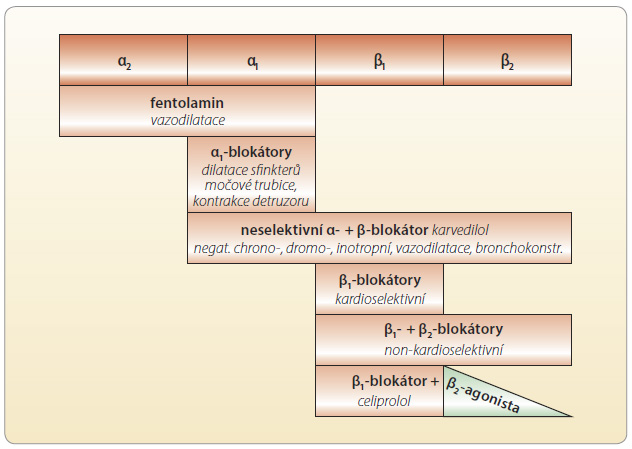 tní odpověď ve vztahu receptor–léčivo může mít různý charakter. Vazba může receptor aktivovat, inhibovat či může současně aktivovat i inhibovat (parciálně agonistický efekt). Příkladem agonistického efektu na úrovni adrenergních receptorů β1 a β2 je adrenalin, příkladem selektivní aktivace receptorů β1 je dobutamin. Obdobně příkladem antagonistického efektu na obou receptorech β1 a β2 jsou neselektivní betablokátory, respektive na receptoru β1 „kardioselektivní“ betablokátory. Parciálními antagonisty jsou pak betablokátory s vnitřní sympatomimetickou aktivitou – např. acebutolol. Při parciálním agonismu mají vysoké dávky léčiva paradoxně menší efekt; nikdy nedosáhneme tak intenzivní inhibice adrenergního systému jako u blokátorů bez vnitřní aktivity. Konečně některé léky mohou jeden adrenergní β-receptor blokovat a druhý stimulovat, příkladem je opět celiprolol, blokátor receptoru β1 a aktivátor receptoru β2. Vzájemný přesah působení agonistů i blokátorů adrenergních receptorů je častý a má klinický význam (obr. 3, 4). Například zmíněný celiprolol je díky aktivaci receptorů β2 výhodnější u astmatiků; karvedilol – díky blokádě receptorů α1 – zase upřednostňujeme v léčbě diabetiků, zachována je citlivost k působení insulinu.
tní odpověď ve vztahu receptor–léčivo může mít různý charakter. Vazba může receptor aktivovat, inhibovat či může současně aktivovat i inhibovat (parciálně agonistický efekt). Příkladem agonistického efektu na úrovni adrenergních receptorů β1 a β2 je adrenalin, příkladem selektivní aktivace receptorů β1 je dobutamin. Obdobně příkladem antagonistického efektu na obou receptorech β1 a β2 jsou neselektivní betablokátory, respektive na receptoru β1 „kardioselektivní“ betablokátory. Parciálními antagonisty jsou pak betablokátory s vnitřní sympatomimetickou aktivitou – např. acebutolol. Při parciálním agonismu mají vysoké dávky léčiva paradoxně menší efekt; nikdy nedosáhneme tak intenzivní inhibice adrenergního systému jako u blokátorů bez vnitřní aktivity. Konečně některé léky mohou jeden adrenergní β-receptor blokovat a druhý stimulovat, příkladem je opět celiprolol, blokátor receptoru β1 a aktivátor receptoru β2. Vzájemný přesah působení agonistů i blokátorů adrenergních receptorů je častý a má klinický význam (obr. 3, 4). Například zmíněný celiprolol je díky aktivaci receptorů β2 výhodnější u astmatiků; karvedilol – díky blokádě receptorů α1 – zase upřednostňujeme v léčbě diabetiků, zachována je citlivost k působení insulinu.
Ovlivnění terapeutické odpovědi na úrovni interakce organismus–léčivo
Výsledný účinek léku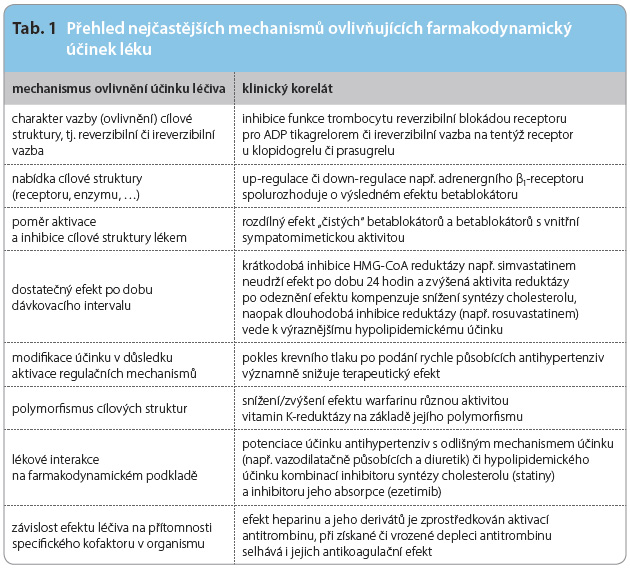 je ovlivňován nejen afinitou k cílové struktuře – k receptoru (a obdobně k enzymu) – a vazbou na ni, ale také řadou okolností nezávislých na vlastnostech léku (tab. 1). Jednou z nich je vlastní exprese receptoru na buněčné membráně (či obdobně aktivita enzymu). Působení léčiva, např. (katecholaminů, betablokátorů apod.) na adrenergní receptor je významně ovlivněno hustotou osazení membrány vlastním receptorem. Exprese receptoru se přitom mění v čase a je ovlivňována i vlastním lékem. Aplikace betablokátoru expresi receptoru na membráně zpravidla zvyšuje, adrenergní stimulace ji naopak snižuje. Up-regulací receptoru β1 v kardiomyocytu po aplikaci např. karvedilolu je vysvětlován relativně velmi malý negativně inotropní efekt léčiva při zachování negativně chronotropního působení. Z vnějších vlivů stimuluje osazení membrány receptorem β1 např. trijodtyronin a tyroxin. Odpověď na adrenergní stimulaci je při tyreotoxikóze výraznější, stejně tak však je potencován efekt betablokátorů. Obdobné vztahy jsou ve variabilní nabídce jednotlivých enzymů, nejen na podkladě genetickém, ale i vlivem řady vnějších faktorů. Podobně efekt léčiva ovlivňuje aktivace regulačních mechanismů. U antihypertenziv s rychlým nástupem účinku vede náhlý pokles krevního tlaku k aktivaci regulačních mechanismů, zejména k adrenergní stimulaci. Ta nejen oslabí výsledný pokles tlaku, ale může vyvolat řadu nežádoucích účinků. Příkladem významu je léčba nifedipinem, kdy rychlý nástup účinku vedl k rychlému a výraznému poklesu tlaku. Ten aktivoval sympatoadrenergní osu a vyplavení katecholaminů vedlo k elektronestabilitě myokardu a zvyšovalo výskyt náhlé smrti na podkladě maligních tachyarytmií. Zavedení blokátorů kalciového kanálu s pomalým nástupem účinku, např. amlodipinu, je proto výrazně výhodnější.
je ovlivňován nejen afinitou k cílové struktuře – k receptoru (a obdobně k enzymu) – a vazbou na ni, ale také řadou okolností nezávislých na vlastnostech léku (tab. 1). Jednou z nich je vlastní exprese receptoru na buněčné membráně (či obdobně aktivita enzymu). Působení léčiva, např. (katecholaminů, betablokátorů apod.) na adrenergní receptor je významně ovlivněno hustotou osazení membrány vlastním receptorem. Exprese receptoru se přitom mění v čase a je ovlivňována i vlastním lékem. Aplikace betablokátoru expresi receptoru na membráně zpravidla zvyšuje, adrenergní stimulace ji naopak snižuje. Up-regulací receptoru β1 v kardiomyocytu po aplikaci např. karvedilolu je vysvětlován relativně velmi malý negativně inotropní efekt léčiva při zachování negativně chronotropního působení. Z vnějších vlivů stimuluje osazení membrány receptorem β1 např. trijodtyronin a tyroxin. Odpověď na adrenergní stimulaci je při tyreotoxikóze výraznější, stejně tak však je potencován efekt betablokátorů. Obdobné vztahy jsou ve variabilní nabídce jednotlivých enzymů, nejen na podkladě genetickém, ale i vlivem řady vnějších faktorů. Podobně efekt léčiva ovlivňuje aktivace regulačních mechanismů. U antihypertenziv s rychlým nástupem účinku vede náhlý pokles krevního tlaku k aktivaci regulačních mechanismů, zejména k adrenergní stimulaci. Ta nejen oslabí výsledný pokles tlaku, ale může vyvolat řadu nežádoucích účinků. Příkladem významu je léčba nifedipinem, kdy rychlý nástup účinku vedl k rychlému a výraznému poklesu tlaku. Ten aktivoval sympatoadrenergní osu a vyplavení katecholaminů vedlo k elektronestabilitě myokardu a zvyšovalo výskyt náhlé smrti na podkladě maligních tachyarytmií. Zavedení blokátorů kalciového kanálu s pomalým nástupem účinku, např. amlodipinu, je proto výrazně výhodnější.
Ovlivnění účinku farmakogenetickými vlivy
Výsledný farmakodynamický efekt je ovlivněn i farmakogenetickou výbavou, tedy polymorfismem cílových struktur působení léku. Většina proteinů v organismu nemá zcela identickou sekvenci aminokyselin mezi jednotlivci; jsou-li tyto drobné odchylky v populaci častější (≥ 1 %), mluvíme o polymorfismu. Totéž se týká bílkovin receptorů, enzymů, iontových kanálů, transportních systémů apod. Většina odchylek neovlivňuje funkci daného proteinu, řada však má biologický význam a přímo zvyšuje či snižuje aktivitu systému, mění odpověď na regulační mechanismy, mění afinitu k přirozeným ligandům či k léčivům apod. Ve farmakologii se setkáváme s plejádou takovýchto odchylek, které modifikují výslednou farmakodynamickou odpověď.
Zůstan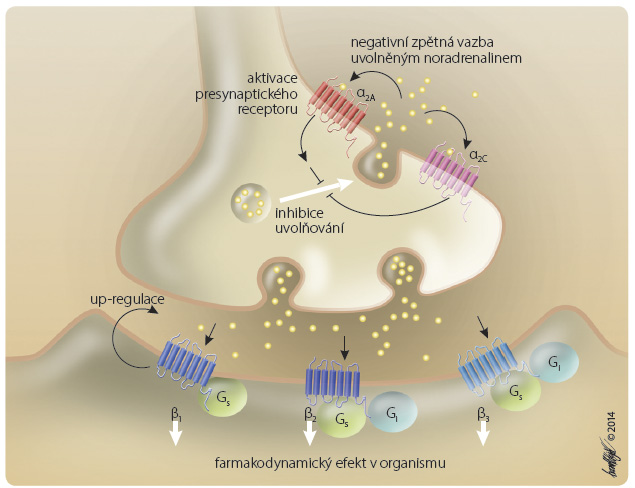 eme-li u příkladů na úrovni adrenergního receptoru β, pak i zde se vyskytuje několik desítek polymorfismů, řada z nich s klinickým významem. Aktivita receptoru β1 je stimulována postsynaptickým receptorem β1, a naopak tlumena aktivací presynaptického receptoru α2 (obr. 5). Nadměrná aktivace receptoru uvolněným noradrenalinem tak výsledný efekt tlumí negativní zpětnou vazbou.
eme-li u příkladů na úrovni adrenergního receptoru β, pak i zde se vyskytuje několik desítek polymorfismů, řada z nich s klinickým významem. Aktivita receptoru β1 je stimulována postsynaptickým receptorem β1, a naopak tlumena aktivací presynaptického receptoru α2 (obr. 5). Nadměrná aktivace receptoru uvolněným noradrenalinem tak výsledný efekt tlumí negativní zpětnou vazbou.
Farmakogenetické vlivy na obou úrovních – presynaptické i postsynaptické – modulují výsledný efekt betablokátorů. Polymorfismus receptoru β1 typu Ser49Gly, který je přítomen asi u 15 % populace, je spojen s výraznější down-regulací receptoru po stimulaci noradrenalinem (s desenzibilizací). U nositelů tohoto genu (poor responders) je pozorována nižší citlivost ke stimulaci i k blokádě receptoru. Naopak ještě častější polymorfismus receptoru α2 Arg389Gly snižuje afinitu noradrenalinu k tomuto presynaptickému inhibičnímu receptoru a několikanásobně zvyšuje citlivost adrenergního nervového zakončení na stimulaci i na blokádu. U většiny nositelů mutací není klinický význam tak velký, jedna „výchylka“ je systémem zpětné vazby kompenzována. Proto musí být „nevýhodných“ polymorfismů více, zpravidla na úrovni pre- i postsynaptického receptoru. Tak například kombinace výskytu dvou polymorfismů – presynaptického receptoru α2c (Del32-325) s nižší inhibiční aktivitou a postsynaptického β1 (Arg389Gly) s vyšší stimulační aktivitou – je spojena s vyšším efektem betablokády při srdečním selhání a snad i s častějším výskytem selhání samotného [1].
Jiným příkladem ovlivnění výsledného dopadu betablokády farmakogenetickou výbavou je kombinace více nepříznivých polymorfismů receptoru β1. Kombinace alel (ADRB1 389Arg/Arg, ADRB1 49Ser/Gly nebo 49 Gly/Gly či dalších) nejenže zvyšovala u nemocných po překonaném infarktu myokardu riziko úmrtí téměř o polovinu (hazard ratio, poměr rizik – HR: 1,49; konfidenční interval – CI: 1,03–2,15; p = 0,033), ale téměř eliminovala příznivý účinek betablokády, respektive u nositelů tohoto rizikového haplotypu byl účinek betablokátorů paradoxně negativní [2]. V jiné indikaci již zmíněné varianty receptoru β1 (ADRB1 389Arg/Arg, či ADRB1 49Ser/Gly) zvyšovaly adjustované riziko výskytu fibrilace síní – při léčbě betablokátory dvojnásobně, bez betablokády dokonce sedminásobně [3].
Farmakogenetické vlivy nepochybně ovlivňují nejen výskyt a průběh chorob, ale též odpověď na léčbu. Obdobná situace je na úrovni receptoru, enzymu či iontového kanálu. Individualizovaná léčba tak bude jedním z nejbližších úkolů, které před námi stojí.
Seznam použité literatury
- [1] Small KM, Wagoner LE, Levin AM, et al. Synergistic polymorphisms of beta1- and alpha2C-adrenergic receptors and the risk of congestive heart failure. N Engl J Med 2002; 347: 1135–1142.
- [2] Matsumoto S, Sakata Y, Nakatani D, et al. Prognostic impact of beta-adrenergic receptor gene polymorphisms on secondary prevention after acute myocardial infarction in the PCI era. JACC 2013; 61: E2123.
- [3] Jeff JM, Donahue BS, Brown-Gentry K, et al. Genetic variation in the beta1-adrenergic receptor is associated with the risk of atrial fibrillation after cardiac surgery. Am Heart J 2014; 167: 101–108.