Bortezomibum
Bortezomib je první inhibitor proteazomu používaný v klinické praxi. Mechanismus jeho protinádorového účinku je zcela odlišný od působení konvenčních protinádorových preparátů. Tento derivát kyseliny boronové vykazuje silnou reverzibilní a specifickou inhibiční aktivitu proti proteazomu, multikatalytickému enzymovému komplexu, který hraje klíčovou úlohu v řízení proteinů kontrolujících vývoj buněčného cyklu a procesy apoptózy. Jeho účinnost byla prokázána v řadě preklinických studií in vitro a in vivo, kdy byl postupně prokázán účinek u buněčných linií myelomu, nehodgkinských maligních lymfomů, nádorů pankreatu, prostaty, ovarií, hlavy a krku či chronické lymfatické leukémie. Kromě vysoké účinnosti a možnosti kombinace s konvenčními léky má příznivý bezpečnostní profil a rychlý nástup účinku často pozorovaný již po prvním cyklu léčby. V současnosti je hlavní indikací mnohočetný myelom, nejčastěji používaným terapeutickým schématem je jednorázová dávka 1,3 mg/m2 podaná 4 dny v 21denním cyklu.
Farmakologická skupina
Bortezomib (původně PS-341, LDP-341, MLN341, JNJ-26866138) vytváří novou farmakologickou skupinu – inhibitor proteazomu. Patří k nové generaci protinádorových léků, jejichž terapeutickým cílem jsou specifické nitrobuněčné procesy v nádorových buňkách kontrolující buněčný cyklus a procesy apoptózy [1]. V anglické literatuře je tento efekt někdy označován jako cílená (targeted) terapie.
Chemické a fyzikální vlastnosti
Bortezomib je modifikovaný dipeptidylborát: [(1R)-3-methyl-1-[[(2S)-1-oxo-3-phenyl-2-[(pyrazinylcarbonyl) amino]propyl]amino]butyl]borát (obr. 1).
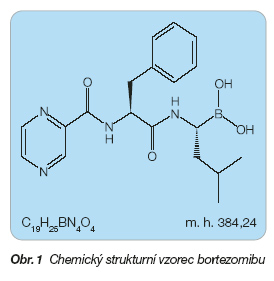
Přípravek obsahuje 3,5 mg bortezomibu v podobě bílého sterilního lyofilizovaného prášku.
Sumární vzorec: C19H25BN4O4
Molekulová hmotnost: 384,24
Rozpustnost bortezomibu jako borátového monomeru ve vodě je 3,3–3,8 mg/ml při pH v rozmezí 2–6,5.
Mechanismus účinku
Bortezomib je selektivním reverzibilním inhibitorem proteazomu. Degradace proteinů v proteazomu je společně s ubikvitinylací (ubikvitinem zprostředkovaná proteazomální degradace) základním biochemickým procesem, který zodpovídá za likvidaci více než 80 % intracelulárních proteinů. Vysoká selektivita umožňuje, aby ubikvitinem zprostředkovaná proteazomální degradace sloužila jako jeden ze základních modulů ovlivňujících biologický poločas řady regulačních proteinů podílejících se na řízení buněčného cyklu, apoptózy, reparace genomové DNA či mezibuněčné komunikace. Pomocí tohoto děje jsou v buňce rovněž eliminovány chybně translatované či nesprávně sestavené proteiny. Pro pochopení mechanismu účinku bortezomibu je nezbytné porozumět procesu eliminace intracelulárních proteinů v proteazomu.
Ubikvitinem zprostředkovaná proteazomální degradace
Ubikvitinem zprostředkovaná proteazomální degradace zahrnuje dva úzce spolu související děje: označení proteinu určeného k eliminaci pomocí jeho kovalentní modifikace polypeptidem – ubikvitinem (ubikvitinylace), a degradaci takto ubikvitinem označeného proteinu ve specializované multikatalytické částici – proteazomu (obr. 2).

Ubikvitin je všudypřítomně se vyskytující polypeptid (8,5 kDa) skládající se ze 76 aminokyselinových zbytků [2]. Pro kovalentní modifikaci degradovaného proteinu je nezbytná aktivace ubikvitinu ubikvitinaktivačním enzymem (E1), jeho následný přenos na ubikvitin-konjugační enzymy (E2), které slouží jako donory aktivovaného ubikvitinu pro ubikvitinylaci cílových proteinů pomocí ubikvitin-ligázové aktivity enzymů E3 [3]. Ubikvitinligázy zprostředkovávají vytvoření kovalentní vazby mezi terminálním glycinem na karboxylovém konci molekuly ubikvitinu a e-aminoskupinou lysinu v proteinu určeném k degradaci [4]. Pro degradaci je obvykle nezbytné navázání většího počtu ubikvitinových molekul na cílový protein, čehož je docilováno řetězením ubikvitinových molekul (polyubikvitinylace). Vysoká specifičnost celého procesu je zajištěna hierarchickým uspořádáním ubikvitinylačních dějů. Na jejich počátku stojí jeden aktivační enzym E1, který přenáší ubikvitin na několik ubikvitin-konjugačních enzymů E2. Ubikvitin-konjugační enzymy jsou schopny interagovat s řadou ze stovek proteinů s ubikvitin-ligázovou aktivitou (E3). K cílovém proteinu je kovalentně vázán v závislosti na charakteru N-koncové aminokyseliny nebo tzv. destrukčního boxu [5].
26S proteazom je bílkovinná partikule skládající se z 20S proteolytické centrální podjednotky a jedné či dvou 19S regulačních podjednotek (obr. 3) [6].
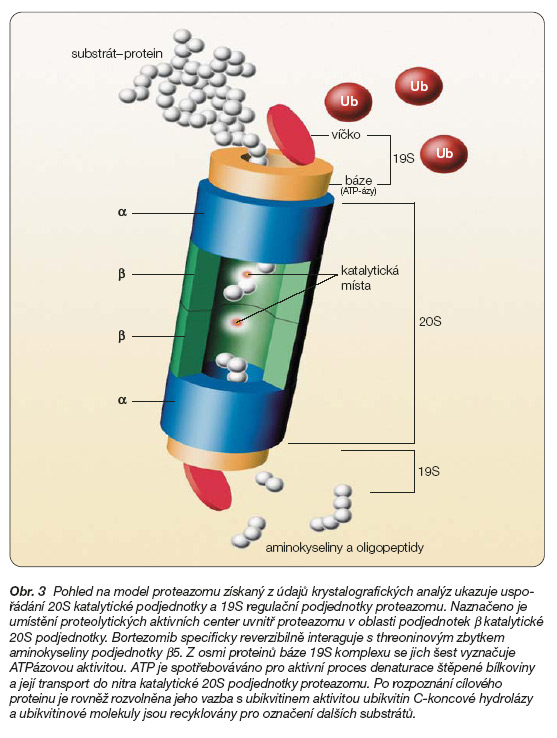
20S katalytická podjednotka proteazomu je tvořena čtveřicí těsně přiléhajících prstenců, z nichž každý sestává ze sedmi různých proteinů. Tyto prstence, označené jako podjednotky a a b, jsou vzájemně orientovány způsobem a-b-b-a, přičemž pouze podjednotky b v centrální části 20S jsou nositeli proteolytické aktivity. Proteolýza probíhá uvnitř dutiny 20S katalytické podjednotky proteazomu a je zprostředkována aktivitou proteázových aktivních center (po třech v každé podjednotce b). Aktivní centra lokalizovaná v oblasti N-konce tří proteinů podjednotek b obsahují threoninový nebo serinový zbytek aminokyseliny, který slouží pro nukleofilní atak karbonylové skupiny peptidové vazby štěpeného proteinu. Na základě podobnosti reakčního mechanismu se známými proteolytickými enzymy jsou aktivní místa v proteazomu označována jako trypsin-like, chymotrypsin-like a postglutamyl-like (též caspase-like). Uvnitř proteazomu je polypeptidový řetězec štěpeného a denaturovaného proteinu postupně degradován na oligopeptidové fragmenty kratší devíti aminokyselinových zbytků.
Na rozdíl od centrální 20S podjednotky je regulační 19S komplex mnohem méně charakterizován [7]. Směrem k centrální 20S podjednotce je lokalizována báze 19S komplexu, ke které přiléhá struktura tvořící víčko (obr. 4).
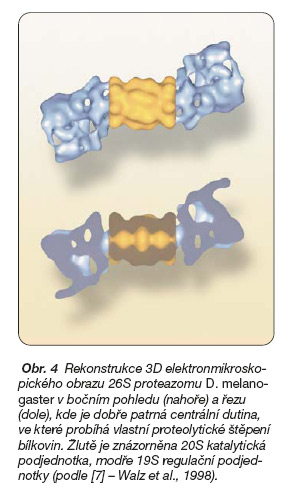
Víčko je tvořeno komplexem složeným z nejméně osmi různých proteinových podjednotek. Proteiny 19S podjednotky se podílejí na rozpoznání degradačního signálu cílového proteinu. Regulace tohoto procesu se účastní i řada dalších bílkovin a bílkovinných komplexů asociovaných s proteazomy [8].
Proteazomy jsou v buňkách hojně zastoupeny. Tvoří přibližně 0,5–1 % všech buněčných proteinů. Vyskytují se nejen v cytoplazmě, ale i v buněčném jádru. Jejich množství je pravděpodobně značně kolísavé a závisí na řadě faktorů, mezi které například patří typ buněk či jejich mitotická aktivita [9].
Proteazomální degradace je zodpovědná za přesně řízenou a správně časovanou likvidaci regulačních proteinů, jako jsou například transkripční faktory, cykliny, inhibitory komplexů cyklinů a cyklin-dependentních kináz, tumor-supresorové geny, inhibitory apoptózy nebo proteiny angažované v reparačních pochodech DNA (tab. 1).

Inhibice proteazomu bortezomibem v léčbě mnohočetného myelomu
Ačkoliv přesný mechanismus působení bortezomibu na nádorové buňky není znám, bylo prokázáno, že nádorové a normální buňky odpovídají na inhibici proteazomu rozdílným způsobem. Zatímco v normálních buňkách způsobuje inhibice proteazomu obvykle zástavu buněčného cyklu, v buňkách nádorových je často jejím výsledkem aktivace apoptózy [10, 11]. Laboratorní výsledky ukázaly signifikantně vyšší citlivost na indukci apoptózy po aplikaci bortezomibu u buněčných linií mnohočetného myelomu než u cirkulujících mononukleárních buněk zdravých osob [12]. Inhibice proteazomu vyvolává stabilizaci řady tumor-supresorových proteinů, včetně p53, p21 nebo p27 [13]. Za kritický faktor působení bortezomibu v buňkách myelomu a dalších hematologických malignit se považuje ovlivnění nukleárního transkripčního faktoru (NF-kB) [12, 14]. NF-kB je heterodimerní transkripční faktor z rodiny Rel (reticuloendotheliosis) kontrolující expresi řady genů, jež stimulují růst buňky, její rezistenci na stresové podněty a proapoptotické faktory, ovlivňují angiogenezi a interakci s buněčným stromatem (obr. 5) [15].
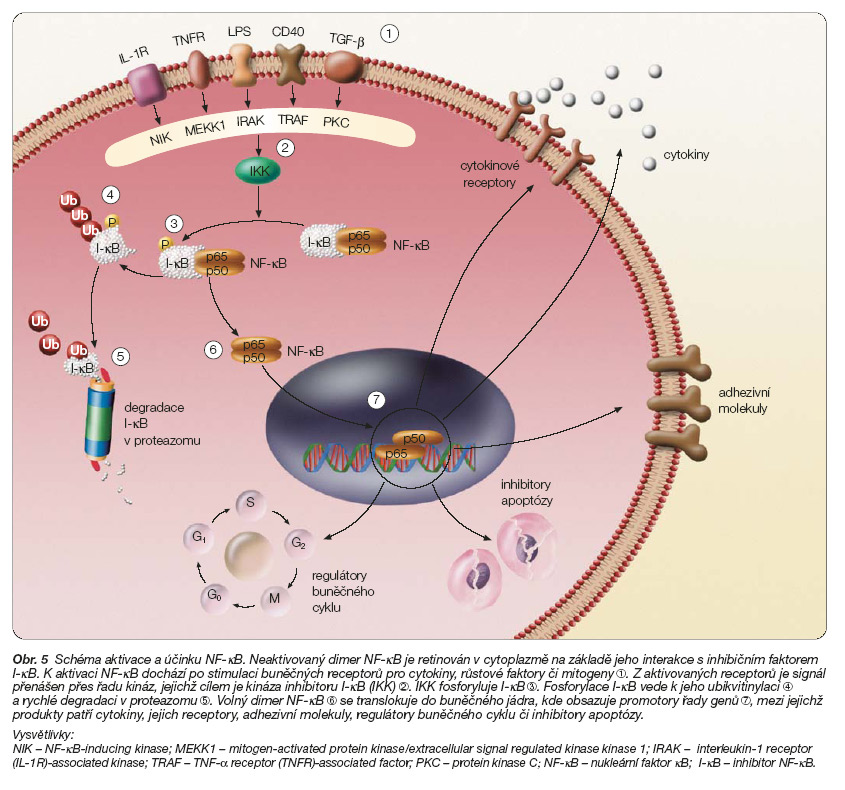
V buňkách mnohočetného myelomu byla prokázána zvýšená exprese NF-kB vyvolávající aktivaci genů pro inhibitory apoptózy Bcl-2, XIAP (X chromosome-linked inhibitor of apoptosis), cIAP-1, cIAP-2 (celular inhibitor of apoptosis protein 1/2), survivin – a sníženou aktivitu genu pro aktivátor apoptózy Bax [16]. Inhibice proteazomu bortezomibem v buňkách mnohočetného myelomu inhibuje rovněž aktivaci NF-kB zprostředkovanou účinky TNF-a (tumor necrosis factor a), která se významným způsobem podílí na interakci nádorových buněk se stromatem v mikroprostředí kostní dřeně.
Farmakodynamické vlastnosti
Bortezomib je silným selektivním inhibitorem chymotrypsin-like aktivity b5-podjednotky 20S proteazomu (Ki = 0,6 nM). Studie in vitro prokázaly jeho výrazný antiproliferační a cytotoxický účinek na řadu nádorových buněčných linií [17]. Tento účinek je navíc unikátní [17]. Inhibice proteazomu bortezomibem je závislá na dávce (graf 1) [11].
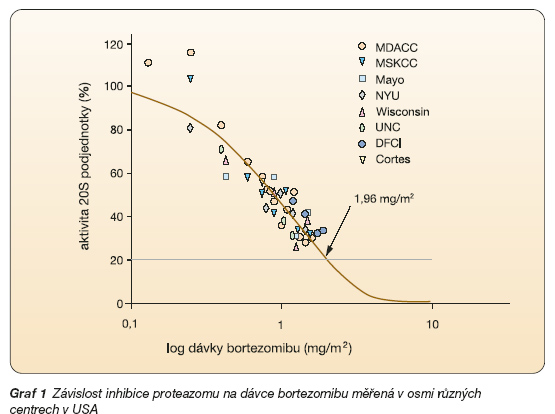
Při podání doporučené dávky 1,3 mg/m2 je inhibice proteazomální enzymatické aktivity přibližně 65 % [18]. Maximální inhibice proteazomu je patrná po 1 hodině od podání; návrat plné proteolytické aktivity proteazomu je pozorovatelný po 72–96 hodinách [19]. Aghajanian a kol. prokázali nezměněnou senzitivitu proteazomu k bortezomibu (stejný inhibiční účinek) 1 hodinu po podání v 1., 4., 8. a 11. dni léčby [18].
Farmakokinetické vlastnosti
Po bolusovém i.v. podání dochází k rychlé redistribuci bortezomibu z plazmy do tkání. Ve studiích na zvířecích modelech bylo prokázáno, že s výjimkou tukové tkáně a některých oblastí CNS se po podání bortezomib dostává do všech tkání [17, 19].
Studie in vitro ukazují, že bortezomib je primárně metabolizován jaterním mikrosomálním systémem cytochromu P-450. Na oxidativním odštěpení borátu z bortezomibu se podílejí izoenzymy 3A4, 2D6, 2C19 a 2C9 [19, 20].
Farmakokinetické studie u nemocných s jaterní nebo renální insuficiencí nejsou ukončeny. V ukončených klinických hodnoceních byli bortezomibem léčeni pacienti s hodnotou clearance kreatininu v rozmezí 13,8–220 ml/min. Nebyla zjištěna souvislost mezi hodnotou clearance kreatininu a inhibiční účinností bortezomibu na proteazom v 1. hodině, četností nežádoucích účinků stupně 3 nebo 4 nebo nutností přerušení léčby. Nemocní se sníženou renální funkcí byli léčeni srovnatelným počtem dávek jako pacienti s normální funkcí ledvin [19].
Základní farmakokinetické charakteristiky bortezomibu udává tab. 2.

Preklinické studie
Na možnost příznivého účinku bortezomibu upozornily výsledky studií in vivo u 60 typů různých humánních tumorů provedené v National Cancer Institute (NCI). Cytotoxický profil byl výjimečný ve srovnání s historickými výsledky u 60 000 jiných látek a odlišný od efektu konvenčních cytostatik [17]. Následně byla účinnost potvrzena řadou autorů v preklinických studiích in vitro a in vivo, kdy byl postupně prokázán účinek u linií myelomu, nehodgkinských lymfomů (NHL), nádorů pankreatu, prostaty, ovarií a hlavy a krku či chronické lymfatické leukémie (CLL) [21, 22].
Bližší analýzy vedly k závěru, že mechanismus účinku u různých typů nádorů se liší a pravděpodobně není závislý na vlivu na jednotlivé proteiny (např. stabilizaci p53, p21, p27), ale k indukci apoptózy dochází zásahem do rovnováhy mezi anti- a proapoptotickými signálními procesy v buňkách [23]. Kromě přímé indukce apoptózy a snížení aktivity NF-kB bylo pozorováno snížení adheze nádorových buněk k buňkám stromatu [24], zásah do nitrobuněčných procesů indukovaných cytokiny [25], senzibilizace vůči apoptóze indukované faktorem TRAIL (TNF-like apoptosis-inducing ligand) [26], nebo naopak porucha protektivního účinku některých antiapoptotických faktorů [27].
První a v blízké budoucnosti zřejmě i jednou z hlavních indikací bortezomibu je mnohočetný myelom, a proto uvádíme podrobněji možné mechanismy účinku pozorované v různých modelech in vitro u buněčných linií myelomových buněk. Bylo prokázáno, že bortezomib přímo indukuje apoptózu myelomových buněk, inhibuje aktivaci NF-kB v nádorových i okolních buňkách, snižuje adherenci myelomových buněk na buňky stromatu, produkci a účinek růstových faktorů myelomové populace – IL-6 (interleukin-6), IGF-1 (insulin-like growth factor-I), VEGF (vascular endothelial growth factor) – a zvyšuje expresi antiapoptotického proteinu Bcl-2. Jeho účinek je nezávislý na buněčném cyklu i stavu hypoxie a není ovlivněn hlavními mechanismy buněčné protinádorové rezistence [23, 28, 29].
Pro klinickou praxi je také velmi významný fakt, že inhibitory proteazomu ovlivňují citlivost nádorových buněk vůči působení cytostatik a radiace. I v tomto případě se jedná pravděpodobně o zásah v několika úrovních [30, 31]. V laboratorních podmínkách byla pozorována 105–106krát vyšší citlivost chemorezistentních buněk mnohočetného myelomu vůči některým cytostatikům – melfalanu, doxorubicinu, mitoxantronu – při kombinaci s necytotoxickými dávkami bortezomibu [32], vzájemná potenciace účinku s dexamethasonem [28] či radioterapií [30]. Na možnost příznivého účinku bortezomibu a chemoterapie (doxorubicin, fluorouracil, cisplatina či paclitaxel) v případě solidních tumorů upozornily např. studie u modelů karcinomu plic [33]. Z hlediska hematologické onkologie lze uvést, že byl pozorován i aditivní efekt u bortezomibu a inhibitorů histondeacetyláz (histone deacetylase inhibitors, HDI) u chronické myeloidní leukémie rezistentní na imatinib mesylát [34].
Klinické zkušenosti
Klinické studie fáze I
Vstupní studie toxicity fáze I prokázala u 9 z 11 zahrnutých nemocných s pokročilým mnohočetným myelomem (MM), kteří dokončili aspoň jeden cyklus léčby, jednu kompletní remisi při dávce 1,04 mg/m2. Osm dalších nemocných dosáhlo minimální léčebné odpovědi či stabilizace onemocnění [35].
Klinické studie fáze II (SUMMIT, CREST)
Studie SUMMIT byla otevřená multicentrická studie fáze II u velmi předléčených nemocných s relabujícím či refrakterním MM, a to nejméně v druhém relapsu onemocnění [36]. Do studie bylo zařazeno 202 nemocných s MM. Dávka bortezomibu byla 1,3 mg/m2 den 1, 4, 8, 11, cyklus byl opakován každé 3 týdny až do dosažení maximálního počtu 8 cyklů. Nemocní s progresí (PG) MM po dvou cyklech léčby dostali dexamethason 20 mg p.o. v den podání a následný den po podání bortezomibu. Podobně byl doplněn do kombinace v případě stabilního onemocnění (SD, stable disease, stabilizace choroby) po 4 cyklech monoterapie bortezomibem.
Pro soubor nemocných byla charakteristická velká předléčenost (medián předchozích léčebných linií = 6; 99,5 % pacientů užívalo dříve steroidy, 92 % alkylační látky, 83 % thalidomid, 81% antracykliny, 64 % autologní transplantaci). Celkem 91 % nemocných bylo refrakterních na poslední podanou léčbu.
Prvním pozorovaným výsledkem byla nezvyklá rychlost léčebného účinku (medián 38 dnů; rozpětí 30–127). Léčebné odpovědi bylo dosaženo ve 35 % případů, s následujícím rozložením: 4 % KR (kompletní remise), 6 % n-KR (téměř KR – jen pozitivní imunofixace), jiná PR (parciální remise) 18 %, MR (minimální odpověď) 7 %. Stabilizace nemoci bylo dosaženo u dalších 24 % pacientů. Léčebná odpověď byla nezávislá na standardních známých prognostických faktorech včetně delece dlouhých ramen chromozomu 13. Rovněž byla nezávislá na typu předchozí léčby [19]. Medián doby do progrese u 67 nemocných s KR, PR a MR byl 12 měsíců, pro celý soubor 7 měsíců. Celkové přežití nemocných bylo 16 měsíců.
Přidání dexamethasonu k bortezomibu u 76 nemocných s PG po druhém nebo v případě SD po čtvrtém cyklu bylo velmi účinné. Ze 74 hodnotitelných nemocných dosáhlo 18 % léčebné odpovědi po přidání kortikoidu, přičemž 6 z 13 nemocných bylo dříve na kortikoidy rezistentních. Toto pozorování bylo předzvěstí úspěchu kombinovaných režimů s bortezomibem využívajících mimořádného synergického účinku bortezomibu s jinými léky bez zkřížení rezistence.
CREST byla druhou studií fáze II u méně předléčených 54 nemocných, zaměřená na ověření optimální dávky [38]. Nemocní byli randomizováni a dostali buď 1,0 mg/ m2, nebo 1,3 mg/m2 ve stejném schématu podání jako ve studii SUMMIT. Bylo zjištěno, že dávka 1,3 mg/m2 je pravděpodobně účinnější, neboť počet celkových léčebných odpovědí byl 50 % proti 33 % při dávce 1,0 mg/m2.
Klinická studie fáze III (APEX)
APEX byla mezinárodní klinická studie srovnávající bortezomib s dexamethasonem u relabujících nemocných s MM [37]. Do studie bylo zařazeno 669 nemocných s MM v 93 centrech, kteří dostali 1–3 léčebné režimy před zařazením do studie a nebyli refrakterní na dexamethason. Při celkovém sledování 8,3 měsíců (medián) lze konstatovat, že bortezomib byl signifikantně více účinný než dexamethason, což se projevilo lepší léčebnou odpovědí (38 % vs 18 %; p = 0,0001), vyšším počtem KR (KR a nKR 13 % vs 2 %) a lepším jednoletým přežitím (80 % vs 66 %; p = 0,0005).
Studie fáze I/II s použitím bortezomibu v kombinacíchs jinými léky
Bortezomib byl do této chvíle zkoušen u nemocných s MM v kombinaci s řadou léků včetně dexamethasonu a prednisonu, melfalanu, doxorubicinu, vincristinu, thalidomidu a dalších. Přehled vybraných režimů u pokročilých i nově diagnostikovaných nemocných s MM viz tab. 3.
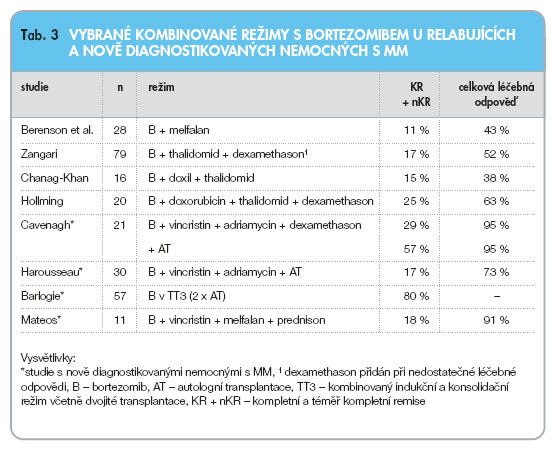
U nemocných s pokročilým MM dosáhly kombinace bortezomibu s melfalanem [39], bortezomibu s thalidomidem [40], bortezomibu s thalidomidem a liposomálním doxorubicinem [41] i kombinace bortezomibu s thalidomidem, dexamethasonem a doxorubicinem [42] pozoruhodných léčebných odpovědí (38–63 % KR + PR). Lze říct, že kombinované režimy vykazovaly zásadně vyšší počet léčebných odpovědí oproti monoterapii bortezomibem včetně vysokého počtu kompletních remisí (11–25 %) srovnatelných jen s léčebnou odpovědí po vysokodávkované léčbě melfalanem s podporou autologního štěpu.
Kombinované režimy použité u nově diagnostikovaných nemocných dosáhly podobných výsledků s dosažením celkové léčebné odpovědi v 71–95 % a velkým počtem kompletních remisí (18 %) v režimech konvenčních [43] i režimech zakončených autologní transplantací [44, 45]. V průběhu prvních studií s podáním bortezomibu před sběrem autologních periferních kmenových krvetvorných buněk bylo sklizeno dostatečné množství CD34+ buněk a následné přihojení štěpu bylo rovněž bezproblémové. Na základě krátkodobých zkušeností lze tedy předpokládat, že bortezomib pravděpodobně nepoškozuje kmenové krvetvorné buňky a může být použit v indukčních režimech před autologní transplantací.
Zařazení do současné palety léčiv
Výsledky preklinických studií, studií fáze I a zejména fáze II vedly ke schválení bortezomibu pro léčbu relabujících a refrakterních forem myelomu, pro druhý a další relaps (tj. jako 3. a další linie léčby) v USA výjimečnou, tzv. zrychlenou procedurou v květnu 2003 [20]. V roce 2004 byl preparát schválen pro klinickou praxi i v zemích Evropské unie, koncem téhož roku MZ ČR schválilo úhradu léku ve stejné indikaci i v naší republice. Na základě prvních jednání je preskripce zatím vázána na doporučení hlavních hematologických center ČR. Počátkem roku 2005 bylo v USA schváleno podání bortezomibu i pro pacienty s prvním relapsem myelomu (tj. pro 2. linii léčby) a doporučeno zahájení zrychlené schvalovací procedury pro nemocné s nehodgkinským lymfomem z plášťové zóny. Od dubna 2005 je použití bortezomibu schváleno pro pacienty s prvním relapsem myelomu (tj. pro 2. linii léčby) i v zemích Evropské unie včetně ČR. Vzhledem k již uvedené skutečnosti, že podstatně vyšší účinnost vykazuje lék v kombinaci s dalšími preparáty, očekávají se výsledky probíhajících studií, které by mohly vést k zařazení bortezomibu do palety léčby myelomu již od jejích úvodních fází. Dále lze očekávat, že se rozšíří i spektrum malignit, u kterých bude bortezomib v blízké budoucnosti používán.
Indikace
V současnosti je hlavní indikací bortezomibu refrakterní/relabující mnohočetný myelom, tj. onemocnění nereagující na předchozí standardní léčbu, kterou je vysokodávkovaná chemoterapie s podporou kmenových hemopoetických buněk (ATKD), resp. konvenční léčba cytostatiky, kortikoidy či thalidomidem. Jak bylo uvedeno, lze v blízké době předpokládat zařazení do palety léčby lymfomů z plášťové zóny, zřejmě také B-chronické lymfatické leukémie a dalších malignit.
Kontraindikace
Přecitlivělost k bortezomibu, bóru nebo jakékoli pomocné látce. Těžké jaterní poškození.
Nežádoucí účinky
V klinických studiích fáze II a III byly hlášeny následující nežádoucí účinky, které mají přinejmenším možný nebo pravděpodobný příčinný vztah k léčbě bortezomibem podle hodnocení zkoušejícího. K velmi častým (frekvence výskytu > 1/10) patří trombocytopenie, anémie, anorexie, periferní neuropatie, bolesti hlavy, závratě, změna chuti, nevolnost, průjem, zvracení, zácpa, vyrážka, bolesti končetin, svalové křeče, bolesti svalů, únava, horečka, slabost. Závažné nežádoucí účinky byly vyhodnoceny maximálně jako časté (frekvence výskytu > 1/100, < 1/10) a patřily k nim trombocytopenie, dehydratace, periferní neuropatie, ortostatická hypotenze, dušnost, dyspeptické poruchy (průjem, zvracení, nevolnost, zácpa), horečka, slabost a únava.
I přes zatím omezené zkušenosti je zřejmé, že hlavními klinicky závažnými komplikacemi terapie bortezomibem jsou trombocytopenie a neuropatie (převážně senzorická, ačkoli byly hlášeny případy motorické neuropatie). Trombocytopenie (výskyt ve 40 % případů ve studii SUMMIT, 30 % ve studii CREST, 60,7 % podle prvních zkušeností v ČR) [46] je však přechodná a ve většině případů se počet krevních destiček (alespoň částečně) upraví do doby zahájení dalšího cyklu. Její příčina není zcela jasná. Z preklinických studií je známo, že bortezomib nemá toxický účinek na hemopoézu včetně megakaryocytů a jejich prekurzorů. Proti přímému myelotoxickému účinku svědčí i krátká doba trvání a častá reverzibilita. Příčinou není zřejmě ani porucha hlavního regulačního mechanismu trombopoézy, neboť u pacientů ve studii SUMMIT byla zjištěna adekvátně zvýšená produkce trombopoetinu [8].
Neurotoxicita byla ve studiích fáze II pozorována u 31 %, resp. 41 % nemocných. V některých případech může vést k předčasnému ukončení léčby. Po vysazení léku však dochází k částečné či úplné úpravě u většiny nemocných (71 %) v relativně krátké době (medián 47 dní) [47].
Úhrnem lze uvést, že většina nežádoucích vedlejších účinků bortezomibu je mírného stupně a klinicky zvládnutelná. V porovnání s ostatními léčebnými možnostmi myelomu je bezpečnostní profil léku příznivý.
Interakce s jinými léčivými přípravky a jiné formy interakce
Cílené studie lékových interakcí nebyly s bortezomibem provedeny. Studie in vitro naznačují, že bortezomib je slabý inhibitor izoenzymů 1A2, 2C9, 2C19, 2D6 a 3A4 cytochromu P-450 (CYP). Na základě omezeného podílu (7 %) izoenzymu CYP 2D6 na metabolismu bortezomibu nelze očekávat, že by slabý metabolický fenotyp CYP 2D6 ovlivnil celkový metabolismus bortezomibu.
V průběhu klinického hodnocení u pacientů s diabetem, kteří užívali perorální antidiabetika, byly hlášeny případy hypoglykémie a hyperglykémie. U pacientů, kteří užívají perorální antidiabetika a jsou léčeni bortezomibem, je nutné pečlivě sledovat hladinu krevního cukru a upravovat dávkování antidiabetik.
Těhotenství a kojení
Klinické údaje o podávání bortezomibu těhotným ženám nejsou k dispozici.
Teratogenní potenciál bortezomibu nebyl plně prozkoumán.V předklinickém hodnocení nevykazoval bortezomib vliv na embryonální fetální vývoj laboratorních potkanů a králíků po podání nejvyšších dávek tolerovaných matkou. Studie se zvířaty zaměřené na průběh porodu a postnatální vývoj nebyly provedeny. Muži i ženy s fertilní kapacitou by měli používat účinná antikoncepční opatření v průběhu léčby a tři měsíce po ukončení léčby bortezomibem. Jestliže je bortezomib podáván během těhotenství nebo pokud žena během léčby tímto lékem otěhotní, je zapotřebí ji seznámit s možnými riziky pro plod.
Není známo, zda je bortezomib vylučován do mateřského mléka. Z důvodu možných závažných nežádoucích účinků pro kojené dítě se matkám doporučuje, aby v průběhu léčby bortezomibem nekojily.
Dávkování a způsob podání
Doporučená úvodní dávka bortezomibu je 1,3 mg/m2 tělesného povrchu dvakrát týdně po dobu dvou týdnů (1., 4., 8. a 11. den) a poté následuje 10denní přestávka (12.–21. den).
Toto 3týdenní období je považováno za léčebný cyklus. Odstup mezi po sobě jdoucími dávkami bortezomibu musí činit nejméně 72 hodin.
Doporučuje se, aby pacienti s potvrzenou kompletní odpovědí podstoupili po ověření remise ještě další dva léčebné cykly s bortezomibem. Doporučuje se rovněž, aby pacienti, kteří odpovídají na léčbu a u kterých nebylo dosaženo kompletní remise, podstoupili celkem 8 léčebných cyklů s bortezomibem.
V současné době je jen omezené množství údajů o opakované léčbě bortezomibem.
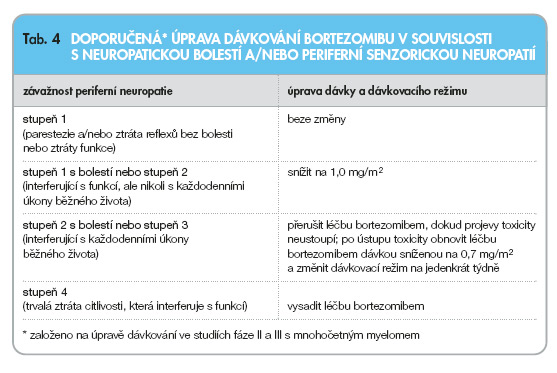
Léčba bortezomibem musí být přerušena při výskytu jakékoli nehematologické toxicity stupně 3 nebo hematologické toxicity stupně 4 s výjimkou neuropatie, jak je uvedeno níže. Jakmile projevy toxicity ustoupí, může být léčba bortezomibem znovu zahájena dávkou sníženou o 25 % (1,3 mg/m2 snížit na 1,0 mg/m2; 1,0 mg/m2 snížit na 0,7 mg/m2). Jestliže toxicita neodezněla nebo se objeví i při nejnižší dávce, musí se uvažovat o vysazení léčby bortezomibem, zejména pokud přínos léčby prokazatelně nepřevýší riziko.
Pacienti, u kterých se v souvislosti s léčbou bortezomibem objevila neuropatická bolest a/nebo periferní neuropatie, mají být léčeni, jak uvádí tab. 4. Pacienti s již existující závažnou neuropatií mohou být léčeni bortezomibem pouze po pečlivém zhodnocení poměru riziko/přínos.
Balení
Viz tab. 5. Jedna injekční lahvička obsahuje 3,5 mg bortezomibu (jako esteru kyseliny boronové s manitolem). Po rekonstituci obsahuje 1 ml injekčního roztoku 1 mg bortezomibu.

Stabilita, podmínky pro skladování
Doba použitelnosti neředěného léku je 2 roky. Rekonstituovaný roztok byl měl být použit neprodleně po přípravě. Chemická a fyzikální stabilita byla prokázána po dobu 8 hodin při teplotě 25 °C při uchovávání v originální injekční lahvičce a/nebo stříkačce před aplikací, s maximální dobou 8 hodin v injekční stříkačce. Lék se skladuje se při teplotě do 30 °C. Injekční lahvičku uchováváme ve vnějším obalu, aby byl přípravek chráněn před světlem.
Lék nesmí být smíchán se žádnými jinými léky.
Seznam použité literatury
- [1] Klener P. Protinádorová chemoterapie pro 21. století. Klin Onkol 2003,16: 243–248.
- [2] Pickart CM, Eddins MJ. Ubiquitin: structures, functions, mechanisms. Biochim Biophys Acta 2004; 1695: 55–72.
- [3] Hershko A. Lessons from the discovery of the ubiquitin system. Trends Biochem Sci 1996; 21: 445–449.
- [4] Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem 1998; 67: 425–479.
- [5] Varshavsky A. The ubiquitin system. Trends Biochem Sci 1997; 22: 383–387.
- [6] Puhler G, Weinkauf S, Bachmann L, Muller S, Engel A, Hegerl R, Baumeister W. Subunit stoichiometry and three-dimensional arrangement in proteasomes from Thermoplasma acidophilum. EMBO J 1992; 11: 1607–1616.
- [7] Walz J, Erdmann A, Kania M, Typke D, Koster AJ, Baumeister W. 26S proteasome structure revealed by three-dimensional electron microscopy. J Struct Biol 1998; 121: 19–29.
- [8] Bajorek M, Glickman MH. Keepers at the final gates: regulatory complexes and gating of the proteasome channel. Cell Mol Life Sci 2004; 61: 1579–1588.
- [9] Wojcik C, DeMartino GN. Intracellular localization of proteasomes. Int J Biochem Cell Biol 2003; 35: 579–589.
- [10] Adams J. Potential for proteasome inhibition in the treatment of cancer. Drug Discov Today 2003; 8: 307–315.
- [11] Adams J. Development of the proteasome inhibitor PS-341. Oncologist 2002; 7: 9–16.
- [12] Richardson PG, Hideshima T, Anderson KC. Bortezomib (PS-341): a novel, first-in-class proteasome inhibitor for the treatment of multiple myeloma and other cancers. Cancer Control 2003; 10: 361–9.
- [13] Kisselev AF, Goldberg AL. Proteasome inhibitors: from research tools to drug candidates. Chem Biol 2001; 8: 739–58.
- [14] Mitchell BS. The proteasome – an emerging therapeutic target in cancer. N Engl J Med 2003; 348: 2597–2598.
- [15] Panwalkar A, Verstovsek S, Giles F. Nuclear factor-kappaB modulation as a therapeutic approach in hematologic malignancies. Cancer 2004; 100: 1578–1589.
- [16] Mitsiades N, Mitsiades CS, Poulaki V, Chauhan D, Richardson PG, Hideshima T, et al. Biologic seque- lae of nuclear factor-kappaB blockade in multiple myeloma: therapeutic applications. Blood 2002; 99: 4079–4086.
- [17] Adams J, Palombella VJ, Sausville EA, Johnson J, Destree A, Lazarus DD, et al. Proteasome inhibitors: a novel class of potent and effective antitumor agents. Cancer Res 1999; 59: 2615–1522.
- [18] Aghajanian C, Soignet S, Dizon DS, Pien CS, Adams J, Elliott PJ, et al. A Phase I Trial of the Novel Proteasome Inhibitor PS341 in Advanced Solid Tumor Malignancies. Clin Cancer Res 2002; 8: 2505–2511.
- [19] Schwartz R, Davidson T. Pharmacology, phar macokinetics, and practical applications of bortezomib. Oncology (Huntingt) 2004; 18:14–21.
- [20] Kane RC, Bross PF, Farrell AT, Pazdur R. Velcade: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist 2003; 8: 508–513.
- [21] Pahler JC, Ruiz S, Niemer I, et al. Effects of the proteazome inhibitor, bortezomib, on apoptosis in isolated lymphocytes obtained from patients with chronic lymphocytic leukemia. Clin Cancer Res 2003; 9 (12): 4570–4577.
- [22] Schenkein D. Proteazome inhibitors in the treatment of B-cell malignancies. Clin Lymphoma 2002; 3 (1):49–55.
- [23] Hideshima T, Mitsiades C, Akiyama M, et al. Molecular mechanism mediating antimyeloma activity of proteazome inhibitor PS-341. Blood 2003; 101 (4): 1530–1534.
- [24] Hideshima T, Richardson P, Chauhan D, et al. The proteazome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res 2001; 61: 3071–3076.
- [25] Hideshima T, Chauhan D, Hayashi T, et al. Proteazome inhibitor PS-341 abrogates IL-6 triggered signaling cascades via caspase-dependent downregulation of gp130 in multiple myeloma. Oncogene 2003; 22 (52): 8386–8393.
- [26] Johnson TR, Stone K, Nikrad M, et al. The proteazome inhibitor PS-341 overcomes TRAIL resistance in Bax and caspase 9-negative or Bcl-xL overexpressing cells. Oncogene 2003;. 22 (32): 4953–4963.
- [27] Feinman R, Pranoti G, Miller S, et al. Proteazome inhibitor PS341 inhibits constitutive NF-B activity and bypasses the anti-apoptotic Bcl-2 signal in human multiple myeloma cells. Blood 2001; 98: 640–643.
- [28] Hideshima T, Chauhan D, Richardson P, et al. NF-B as a therapeutic target in multiple myeloma. Biol Chem 2002; 277 (19): 16639–16647.
- [29] Richardson P. Clinical update: proteazome inhibitors in hematologic malignancies. Cancer Treat Rev 2003; 29 (Suppl 1): 33–39.
- [30] Mitsiades N, Mitsiades CS, Richardson PG, et al. The proteazome inhibitor PS-341 potentiates sensitivity of multiple myeloma cells to conventional chemotherapeutic agents: therapeutic applications. Blood 2003; 101 (6): 2377–2380.
- [31] Wang CY, Cusack JC, Jr, Liu R, Baldwin AS, Jr. Control of inducible chemoresistance: enhanced anti-tumor therapy through increased apoptosis by inhibition of NF-B. Nat Med 1999; 5: 412–417.
- [32] Ma MH, Yang HH, Parker K, et al. The proteazome inhibitor PS-341 markedly enhances sensitivity of multiple myeloma tumor cells to chemotherapeutic agents. Clin Cancer Res 2003; 9: 1136–1144.
- [33] Adams J, Palombella VJ, Elliott P. J. Proteazome inhibition: a new strategy in cancer treatment. Investig New Drugs 2000; 18: 109–121.
- [34] Yu C, Rahmani M, Conrad D, et al. The proteazome inhibitor bortezomib interacts synergistically with histone deacetylase inhibitors to induce apoptosis in Bcr/Abl+ cells sensitive and resistant to STI571. Blood 2003; 102 (10): 3765–3774.
- [35] Orlowski RZ, Stinchcombe TE, Mitchell BS, et al. Phase I trial of the proteazome inhibitor PS-341 in patients with refractory hematologic malignancies. J Clin Oncol 2002; 20: 4420–4427.
- [36] Richardson PG, Barlogie B, Berenson J, et al. A phase 2 study of bortezomib in relapsed refractory myeloma. N Eng J Med 2003; 348: 2609–2617.
- [37] Richardson PG, Sonneveld P, Schuster M, et al. Bortezomib demonstrates superior efficacy to high-dose dexamethasone in relapsed multiple myeloma: final report of the APEX study. Blood 2004; (abstr. 336.5).
- [38] Jagannath S, Barlogie B, Berenson J, et al. A phase 2 study of two doses of bortezomib in relapsed and refractory myeloma. Br J Haematol 2004; 127: 165–172.
- [39] Berenson J, Yang H, Swift R, et al. Bortezomib in combination with melphalan in the treatment of relapsed or refractory multiple myeloma: a phase I/II study. Blood 2004; 104: (abstr. 209): 64a.
- [40] Zangari M, Barlogie B, Hollming K, et al. Marked activity of Velcade plus thalidomide (V + T) in advanced and refractory multiple myeloma (MM). Blood 2004; 104 (abstr. 1480): 413a.
- [41] Chanag-Khan AA, Miller KC, McCarthy P, et al. A phase II study of Velcade (V), Doxil (D) in combination with low-dose thalidomide (T) as salvage therapy for patients with relapsed (rel) or refractory (ref) multiple myeloma (MM) and Waldenström Macroglobulinemia (WM): encour-aging preliminary results. Blood 2004; 104 (abstr. 2421): 665a.
- [42] Hollming K, Stover J, Talamo G, et al. Bortezomib (Velcade) + Adriamycin = thalidomide + dexamethasone (VADT) as an effective regimen in patients with refractory or relapsed multiple myeloma (MM). Blood 2004; 104 (abstr. 2399): 659 a.
- [43] Mateos MV, Bladé J, Mediavilla JD, et al. A phase I/II national multicenter, open-label study of bortezomib plus melphalan and prednison (V-MP) in elderly untreated multiple myeloma patients. Blood 2004; 104: (abstr. 3462): 943a.
- [44] Cavenagh JD, Curry N, Stec J, et al. PAD Combination Therapy (Bortezomib/Adriamycin and Dexamethasone) for Untreated Multiple Myeloma. Journal of Clinical Oncology 2004; 22: (July 15 Supplement, abstr. 6550): 14 S.
- [45] Harousseau JL. New strategies in transplantation therapy. Oncology (Huntingt) 2004; 18: 1614–1615, 1617.
- [46] Špička I, Hájek R, Vytřasová M, et al. Inhibitor proteazomu – bortezomib (Velcade) – v léčbě refrakterního mnohočetného myelomu. První zkušenosti v ČR. ČLč, v tisku.
- [47] Richardson P, Briemberg H, Jagannath S, et al. Characterization and reversibility of perifpheral neuropathy in patients with advanced multiple myeloma treated with bortezomib (Velcade). The SUMMIT and CREST study group. Innovations in cancer: Advances in therapy with proteazome inhibition and supportive care. Valencia, Sept. 2004, abstr.