Kladribin ‒ nová alternativa v léčbě vysoce aktivní relabující‑remitující roztroušené sklerózy
Key words: multiple sclerosis, cladribine, deoxyadenosine, intermittent immunosuppression.
Úvod
Kladribin je léčivem původně vyvíjeným a registrovaným v indikaci trichocelulární leukemie. První zmínky o možnosti jeho využití pro léčbu roztroušené sklerózy (RS) se datují od poloviny devadesátých let minulého století, a to zpočátku v indikaci chronicky progredující formy RS parenterálním podáním [1].
První multicentrickou, dvojitě zaslepenou, placebem kontrolovanou studií fáze III v indikaci relabující remitující RS (RR RS) bylo klinické hodnocení s názvem CLARITY, jehož výsledky byly publikovány v roce 2010 [2]. Perorální kladribin byl následně schválen pro léčbu RS v Rusku a v Austrálii. Vzhledem k výskytu novotvarů v léčebné větvi vyjádřila Evropská léková agentura (EMA) obavy ze zavedení přípravku do léčebné praxe. Ve studii CLARITY totiž ve skupině s placebem nebyl zachycen žádný případ malignit, kdežto ve skupinách pacientů léčených kladribinem se vyskytlo celkem 10 novotvarů. Evropská léková agentura registraci pozastavila a uvedla, že si přeje provést další bezpečnostní analýzy nebo jiné studie, než přistoupí k posouzení nové žádosti o schválení. Výrobce tedy žádost o registraci stáhl a rozhodl se pokračovat v klinických studiích. Navíc byl zaveden registr PREMIERE (Prospective Observational Long term Safety Registry of Multiple Sclerosis Patients Who Have Participated in Cladribine Clinical Studies) shromažďující bezpečnostní data pacientů, kteří se účastnili příslušných studií.
Ve studii ORACLE MS, publikované o čtyři roky později, bylo referováno 0,48 % výskytu novotvarů ve skupině nemocných léčených kladribinem oproti 2,91 % ve skupině s placebem [3]. Tyto nálezy byly podobné jako u jiného přípravku používaného v léčbě RS ‒ fingolimodu, a to ve studii FREEDOMS (fingolimod 0,9 %, placebo 2,4 %) [4]. Frekvence výskytu malignit byla dále zkoumána v metaanalýze 11 studií fáze III u léků ovlivňujících průběh onemocnění (disease modifying drugs, DMDs) [5]. Celková míra malignit v placebem kontrolovaných studiích byla 0,6 % pro ramena s DMDs a významně se nelišila od výskytu novotvarů 0,34 % hlášeného při podávání kladribinu ve studii CLARITY a od výskytu 0,49 % u kladribinu ve studii ORACLE MS. Co se lišilo, byl výskyt malignit ve skupinách s placebem ‒ celkově 1,1 % u studií s DMDs ve srovnání s 0 % ve studii CLARITY. Autoři analýzy dospěli k závěru, že údaje nemohou potvrdit, že v případě kladribinu existuje nadměrné riziko vzniku malignity. Další bezpečnostní informace z registru PREMIERE byly prezentovány na výročním zasedání Americké neurologické akademie (AAN) v roce 2017 [6]. Registr shromáždil údaje od 923 pacientů, kteří dostávali perorální dávku kladribinu 3,5 mg/kg (3 433 pacientoroků). Incidence novotvarů byla 1,14 na 100 pacientoroků u kladribinu v dávce 3,5 mg/kg ve srovnání s 1,01/100 pacientoroků pro placebo. Nebyly pozorovány žádné hematologické malignity obvykle spojené s imunosupresí. Tyto skutečnosti byly natolik přesvědčivé, že EMA dospěla k názoru, že perorální kladribin má přijatelný bezpečnostní profil, takže v srpnu 2017 došlo k jeho definitivní registraci.
Farmakoterapeutická skupina a mechanismus účinku
Kladribin je podle nové klasifikace z roku 2017 řazen mezi Selektivní imunosupresiva, ATC kód L04AA40.
Jedná se o nukleosidový analog deoxyadenosinu (2 CdA)
(obr. 1). Do 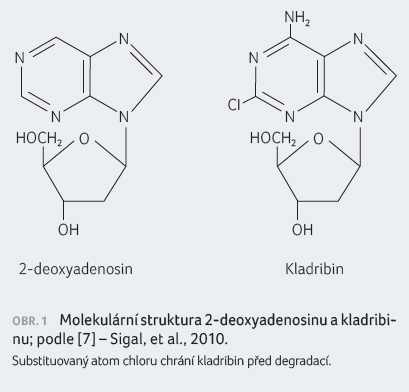 buňky
vstupuje prostřednictvím účinných transportních systémů.
Substituce chloru v purinovém kruhu chrání kladribin před
degradací adenosindeaminázou (ADA), což zvyšuje dobu setrvání
proléčiva kladribinu v intracelulárním prostoru. Následné
fosforylace kladribinu na jeho aktivní trifosfátovou formu,
2 chlorodeoxyadenosin trifosfát (Cd ATP), je zvláště
účinně dosaženo v lymfocytech v důsledku jejich
konstitučně vysoké koncentrace deoxycytidinkinázy (DCK)
a relativně nízké koncentrace 5’ nukleotidázy (5’
NT). Vysoký poměr DCK k 5’ NT preferuje akumulaci
Cd ATP, což zvyšuje náchylnost lymfocytů k buněčné
smrti. V důsledku nižšího poměru DCK/5’ NT jsou
další buňky odvozené z kostní dřeně méně postižené
než lymfocyty. Deoxycytidinkináza je enzym limitující míru
konverze proléčiva kladribinu na jeho aktivní trifosfátovou
formu, což vede k selektivní depleci dělících se
a nedělících se T a B lymfocytů.
buňky
vstupuje prostřednictvím účinných transportních systémů.
Substituce chloru v purinovém kruhu chrání kladribin před
degradací adenosindeaminázou (ADA), což zvyšuje dobu setrvání
proléčiva kladribinu v intracelulárním prostoru. Následné
fosforylace kladribinu na jeho aktivní trifosfátovou formu,
2 chlorodeoxyadenosin trifosfát (Cd ATP), je zvláště
účinně dosaženo v lymfocytech v důsledku jejich
konstitučně vysoké koncentrace deoxycytidinkinázy (DCK)
a relativně nízké koncentrace 5’ nukleotidázy (5’
NT). Vysoký poměr DCK k 5’ NT preferuje akumulaci
Cd ATP, což zvyšuje náchylnost lymfocytů k buněčné
smrti. V důsledku nižšího poměru DCK/5’ NT jsou
další buňky odvozené z kostní dřeně méně postižené
než lymfocyty. Deoxycytidinkináza je enzym limitující míru
konverze proléčiva kladribinu na jeho aktivní trifosfátovou
formu, což vede k selektivní depleci dělících se
a nedělících se T a B lymfocytů.
Primární mechanismus účinku Cd ATP indukující apoptózu
má přímé a nepřímé účinky na syntézu DNA a na
funkci mitochondrií. V dělících se buňkách interferuje
Cd ATP se syntézou DNA prostřednictvím inhibice
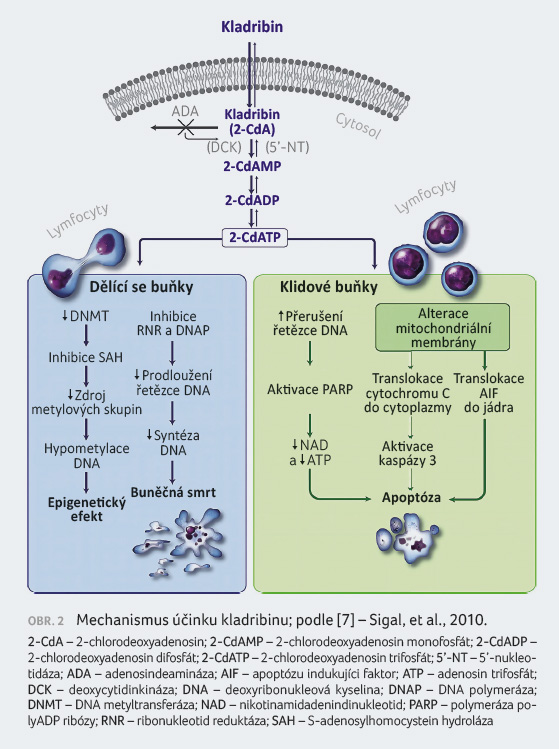 ribonukleotidreduktázy a soutěží s deoxyadenosin
trifosfátem o inkorporaci do DNA působením DNA
polymeráz. V klidových buňkách působí kladribin zlomy
jednoduché šroubovice DNA, rychlé spotřebování
nikotinamid adenin dinukleotidu, depleci ATP a buněčnou
smrt. Existují důkazy, že kladribin může také způsobit
apoptózu závislou a nezávislou na kaspáze
prostřednictvím uvolnění cytochromu C a faktoru indukujícího
apoptózu do cytosolu nedělících se buněk [7,8].
Podrobnější mechanismus účinku kladribinu je znázorněn
na obrázku 2.
ribonukleotidreduktázy a soutěží s deoxyadenosin
trifosfátem o inkorporaci do DNA působením DNA
polymeráz. V klidových buňkách působí kladribin zlomy
jednoduché šroubovice DNA, rychlé spotřebování
nikotinamid adenin dinukleotidu, depleci ATP a buněčnou
smrt. Existují důkazy, že kladribin může také způsobit
apoptózu závislou a nezávislou na kaspáze
prostřednictvím uvolnění cytochromu C a faktoru indukujícího
apoptózu do cytosolu nedělících se buněk [7,8].
Podrobnější mechanismus účinku kladribinu je znázorněn
na obrázku 2.
V imunopatogenezi RS hrají klíčovou roli různé typy imunitních buněk, včetně autoreaktivních T a B lymfocytů. Mechanismus terapeutického účinku kladribinu u RS není plně objasněn, ale předpokládá se, že jeho predominantní účinek na T a B lymfocyty přerušuje kaskádu imunitních pochodů, které mají u RS hlavní význam. Změny míry exprese DCK a 5’ NT mezi podtypy imunitních buněk mohou vysvětlit rozdíly v citlivosti imunitních buněk na kladribin. Z důvodu těchto hodnot míry exprese jsou buňky vrozeného imunitního systému ovlivněny méně než buňky adaptivního imunitního systému [9].
Farmakodynamické účinky
Kladribin má dlouhotrvající účinky vyvolané preferenčním zacílením na lymfocyty a autoimunitní procesy zapojené do patofyziologie RS. V klinických studiích byl největší podíl pacientů s lymfopenií stupně 3 nebo 4 (< 500‒200 buněk/mm³ nebo < 200 buněk/mm³) pozorován dva měsíce po první dávce kladribinu každý rok, což ukazuje časovou prodlevu mezi plazmatickými koncentracemi kladribinu a maximálním hematologickým účinkem. Ve studiích ukazují údaje s navrženou kumulativní dávkou 3,5 mg/kg tělesné hmotnosti postupné zlepšení středního počtu lymfocytů zpět na normální rozmezí v 84. týdnu od podání první dávky kladribinu (přibližně 30 týdnů po poslední dávce kladribinu). Počet lymfocytů u více než 75 % pacientů se vrátil do normálního rozmezí do 144. týdne od první dávky kladribinu (přibližně 90 týdnů po poslední dávce kladribinu). Léčba perorálním kladribinem vede k rychlému snížení koncentrace cirkulujících CD4+ a CD8+ T lymfocytů. Přitom CD8+ T lymfocyty vykazují méně výrazné snížení koncentrace a rychlejší zotavení než CD4+ T lymfocyty, což vede k dočasnému snížení poměru CD4 a CD8. Kladribin snižuje počet CD19+ B lymfocytů a počet CD16+/CD56+ NK buněk (tzv. natural killers), které se také zotavují rychleji než CD4+ T lymfocyty. Kladribin je v současné době jediným zástupcem specifické skupiny léků umožňujících tzv. selektivní imunorekonstituční terapii (selective immune reconstitution therapy, SIRT). Tato léčba spočívá v pulzní imunoterapii vedoucí k dlouhodobým změnám imunitních funkcí. Imunosuprese je v případě SIRT pouze krátkodobá, po každém léčebném pulzu dochází alespoň k částečné obnově počtu lymfocytů [10,11].
Farmakokinetické vlastnosti
Kladribin je proléčivo, které musí být intracelulárně fosforylováno, aby bylo biologicky účinné. Farmakokinetické vlastnosti kladribinu byly studovány po perorálním a intravenózním podání u pacientů s RS a u pacientů s malignitami a v systémech in vitro. Nebyly provedeny žádné studie hodnotící farmakokinetické vlastnosti kladribinu u starších nebo pediatrických pacientů s RS nebo u pacientů s poruchou funkce ledvin nebo jater. Populační kinetická analýza neprokázala vliv věku (v rozmezí od 18 do 65 let) nebo pohlaví na farmakokinetické vlastnosti kladribinu.
Absorpce
Po perorálním podání se kladribin rychle absorbuje. Podání 10 mg kladribinu mělo za následek průměrné hodnoty maximální plazmatické koncentrace (cmax) kladribinu v rozmezí 22‒29 ng/ml a odpovídající průměrné hodnoty plochy pod křivkou sérové koncentrace (AUC) v rozmezí 80‒101 ng·h/ml (aritmetické průměry z různých studií). Když byl perorální kladribin podáván nalačno, byl medián času dosažení maximální plazmatické koncentrace (tmax) 0,5 hodiny (rozmezí 0,5‒1,5 h). Když byl přípravek podán s jídlem s vysokým obsahem tuku, absorpce kladribinu se zpozdila (medián tmax 1,5 h, rozmezí 1‒3 h) a hodnota cmax se snížila o 29 % (na základě geometrického průměru), zatímco hodnota AUC zůstala nezměněná. Biologická dostupnost 10 mg perorálního kladribinu byla přibližně 40 %.
Po perorálním podání kladribinu v dávkách 3–20 mg se hodnoty cmax a AUC zvýšily způsobem úměrným dávce, což naznačuje, že absorpce perorální dávky až do 20 mg není ovlivněna rychlostí nebo kapacitou omezenými procesy. Po opakovaném podávání nebyla pozorována žádná významná kumulace léčiva v plazmě. Nic neukazuje na to, že by se farmakokinetika kladribinu mohla měnit v závislosti na čase po opakovaném podávání.
Distribuce
Distribuční objem je značný, což ukazuje na rozsáhlou distribuci v tkáních a na intracelulární vychytávání. Studie odhalily průměrný distribuční objem kladribinu v rozmezí 480‒490 litrů. Dvacet procent kladribinu se váže na plazmatické proteiny nezávisle na plazmatické koncentraci.
Distribuce kladribinu přes biologické membrány je podporována různými transportními proteiny, včetně ENT1, CNT3 a BCRP. In vitro studie ukazují, že eflux kladribinu souvisí s P glykoproteinem (P gp) jen minimálně. Klinicky významné interakce s inhibitory P gp se neočekávají. Možné důsledky indukce P gp na biologickou dostupnost kladribinu dosud nebyly hodnoceny. In vitro studie ukázaly zanedbatelné vychytávání kladribinu zprostředkované transportéry do lidských hepatocytů.
Kladribin má potenciál pronikat hematoencefalickou bariérou. V malé studii u pacientů s karcinomem byl prokázán poměr koncentrací v mozkomíšním moku/plazmě přibližně 0,25. Kladribin a/nebo jeho fosforylované metabolity se ve značné míře akumulují a zůstávají v lidských lymfocytech. In vitro byl zjištěn intracelulární vs. extracelulární poměr v hodnotě kolem 30‒40 již jednu hodinu po expozici kladribinu.
Biotransformace a eliminace
Metabolismus kladribinu byl zkoumán u pacientů s RS po perorálním podání jedné 10mg tablety a jedné 3mg intravenózně podané dávky. Jak po perorálním, tak i po intravenózním podání byla výchozí sloučenina kladribin hlavní složkou přítomnou v plazmě a moči. Metabolit 2 chloroadenin byl méně důležitým metabolitem v plazmě i v moči, představoval tedy pouze ≤ 3 % plazmatické expozice původnímu léčivu po perorálním podání. Z jiných metabolitů se v plazmě a v moči nacházejí pouze stopy. V hepatálních systémech in vitro byl pozorován zanedbatelný metabolismus kladribinu (minimálně 90 % byla nezměněná léčivá látka). Kladribin není relevantní substrát enzymů cytochromu P450 a nevykazuje významný potenciál účinku jako inhibitor CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 a CYP3A4. Neočekává se, že inhibice těchto enzymů nebo genetické polymorfismy (např. CYP2D6, CYP2C9 nebo CYP2C19) povedou ke klinicky významným účinkům na farmakokinetiku nebo expozici kladribinu. Léčivo nemá žádný klinicky významný indukční účinek na enzymy CYP1A2, CYP2B6 a CYP3A4. Po vstupu do cílových buněk je kladribin fosforylován na kladribin monofosfát (Cd AMP) pomocí DCK (a v mitochondriích také pomocí deoxyguanosinkinázy); Cd AMP je dále fosforylován na kladribin difosfát (Cd ADP) a kladribin trifosfát (Cd ATP). Defosforylace a deaktivace Cd AMP je katalyzována cytoplazmatickou 5’ NT. Ve studii intracelulárních farmakokinetických vlastností Cd AMP a Cd ATP u pacientů s chronickou myeloidní leukemií byla koncentrace Cd ATP přibližně poloviční v porovnání s koncentrací Cd AMP. Intracelulární poločas Cd AMP byl 15 hodin, intracelulární poločas Cd ATP byl 10 hodin.
Na základě sdružených populačních farmakokinetických údajů z různých studií byly střední hodnoty eliminace 22,2 l/h pro renální clearance a 23,4 l/h pro nerenální clearance. Renální clearance překročila rychlost glomerulární filtrace, což ukazuje na aktivní tubulární sekreci kladribinu. Nerenální část eliminace kladribinu (přibližně 50 %) sestává ze zanedbatelného hepatálního metabolismu a z rozsáhlé intracelulární distribuce a zachycování aktivního kladribinu (Cd ATP) uvnitř cílového intracelulárního kompartmentu (tj. lymfocyty) a z následné eliminace intracelulárního Cd ATP podle životního pulzu a eliminačních mechanismů těchto buněk. Odhadovaný terminální poločas u typického pacienta z populační farmakokinetické analýzy je přibližně jeden den. To však nevede k žádné akumulaci léku po dávkování jednou denně, protože tento eliminační poločas představuje pouze malou část AUC [12,13,21].
Klinické zkušenosti
Účinnost a bezpečnost kladribinu byla hodnocena v randomizované, dvojitě zaslepené, placebem kontrolované studii (CLARITY) u 1 326 pacientů s RR RS [2]. Pacienti museli prodělat nejméně jeden relaps během předchozích 12 měsíců. V celkové populaci studie byl průměrný věk subjektů 39 let (rozmezí 18‒65 let) a poměr žen a mužů byl asi 2 : 1. Průměrná doba trvání onemocnění RS před zařazením do studie byla 8,7 roku a medián počátečních hodnot neurologického postižení hodnoceného na základě skóre Kurtzkeho stupnice postižení (Expanded Disability Status Scale, EDSS) ve všech léčebných skupinách byl 3,0 (rozmezí 0‒6,0). Více než dvě třetiny pacientů ve studii nebyly nikdy předtím léčeny DMDs. Zbývající pacienti byli předléčeni interferonem beta 1a, interferonem beta 1b, glatiramer acetátem nebo natalizumabem.
Primárním cílem studie bylo vyhodnotit účinnost kladribinu
oproti placebu ve snížení roční míry relapsů (annualised
relapse rate, ARR), ve zpomalení progrese invalidity a ve
snížení počtu aktivních lézí měřených pomocí magnetické
rezonance (MR).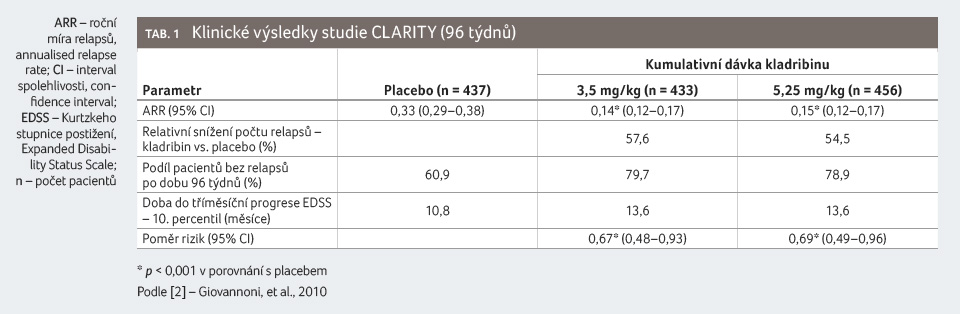
Pacienti dostávali buď placebo (n = 437), nebo kumulativní dávku kladribinu 3,5 mg/kg (n = 433) či 5,25 mg/kg tělesné hmotnosti (n = 456) v průběhu 96týdenní (dvouleté) fáze studie ve dvou léčebných pulzech. Pacienti randomizovaní do skupiny kumulativní dávky 3,5 mg/kg dostali první léčebný pulz v 1. a 5. týdnu v prvním roce a druhý léčebný pulz v 1. a 5. týdnu ve druhém roce. Pacienti randomizovaní do skupiny kumulativní dávky 5,25 mg/kg dostali další léčbu v 9. a 13. týdnu v prvním roce. Většina nemocných v léčebných skupinách placeba (87,0 %), kladribinu 3,5 mg/kg (91,9 %) a kladribinu 5,25 mg/kg (89,0 %) dokončila plných 96 týdnů studie.
Pacienti s RR RS léčení kladribinem v dávce
3,5 mg/kg vykázali statisticky významná zlepšení parametru
ARR, podílu nemocných bez relapsu po dobu 96 týdnů, podílu
nemocných bez trvalé invalidity po dobu 96 týdnů a doby
do tříměsíční progrese EDSS v porovnání s pacienty
v placebové skupině. Výsledky jsou patrné z tabulky 1 a grafů 1, 2, 3, 4.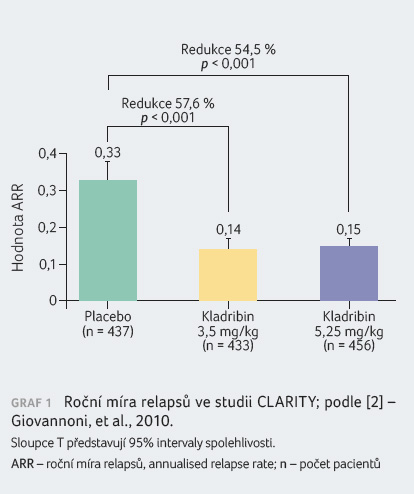
U léčebné skupiny s kladribinem v dávce
3,5 mg/kg v porovnání se skupinou s placebem došlo
ke statisticky významnému snížení výskytu gadolinium
enhancujících (Gd+) T1 lézí, aktivních T2 lézí
a kombinovaných unikátních lézí, jak bylo prokázáno na MR
mozku po celou dobu 96týdenní studie. V porovnání se
skupinou s placebem vykázali pacienti léčení kladribinem
relativní snížení o 86 % v průměrném počtu Gd+ T1
lézí (upravený průměrný počet pro skupiny kladribinu v dávce
3,5 mg/kg a placeba byl 0,12 a 0,91, v uvedeném
pořadí), relativní snížení o 73 % v průměrném
počtu aktivních T2 lézí (upravený průměrný počet pro skupiny
kladribinu v dávce 3,5 mg/kg a placeba byl 0,38 a 1,43,
v uvedeném pořadí) a relativní snížení o 74 %
v průměrném počtu kombinovaných unikátních lézí
na pacienta a snímek (upravený průměrný počet pro
skupiny kladribinu v dávce 3,5 mg/kg a placeba byl 0,43
a 1,72, v uvedeném pořadí; p < 0,001
pro všechny tři cílové ukazatele na MR).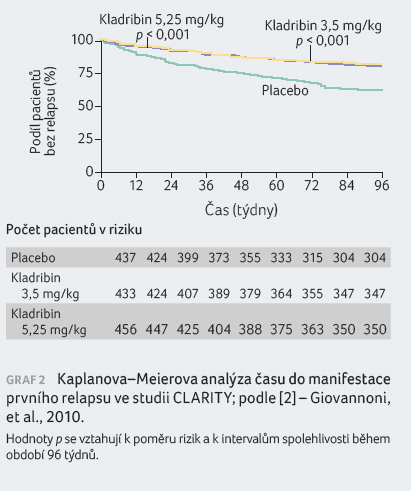
Analýza studie CLARITY hodnotila roční procentuální změnu objemu mozkové tkáně. Ta byla v případě placeba ‒0,70 % ± 0,79, u pacientů léčených kladribinem v dávce 3,5 mg/kg ‒0,56 % ± 0,68 (p = 0,010), u pacientů s terapií dávkou 5,25 mg/kg představovala ‒0,57 % ± 0,72 (p = 0,019). Kladribin tedy statisticky významně redukoval mozkovou atrofii ve srovnání s placebem [14].
Post hoc analýza
doby do šestiměsíční potvrzené progrese EDSS vedla k 47%
snížení rizika progrese invalidity u skupiny s kladribinem
v dávce 3,5 mg/kg v porovnání s placebem (OR
[poměr šancí] 0,53; 95% CI [interval spolehlivosti] 0,36‒0,79;
p < 0,05);
ve skupině s placebem bylo dosaženo 10. percentilu
za 245 dnů a ve skupině s kladribinem v dávce
3,5 mg/kg jej nebylo během studie dosaženo vůbec. Vyšší
kumulativní dávka neměla žádný klinicky významný přínos,
souvisela ale s vyšší incidencí lymfopenie stupně vyššího
než 3 (44,9 % ve skupině s kladribinem v dávce
5,25 mg/kg versus 25,6 % ve skupině s kladribinem
v dávce 3,5 mg/kg)
[15].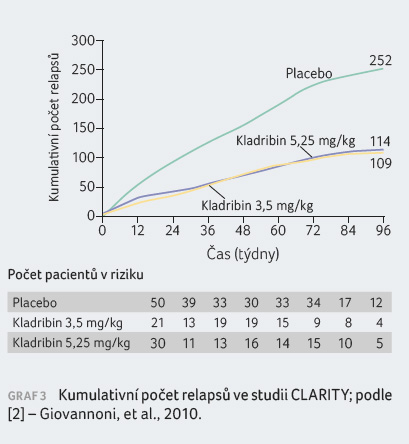
Z počtu 1 326 pacientů, kteří byli randomizováni
ve studii CLARITY, bylo 1 192 hodnoceno z hlediska
bezpříznakového stavu po dobu 96 týdnů. V průběhu 24
týdnů bylo 266 (67 %) z 395 pacientů léčených
kladribinem v dávce 3,5 mg/kg a 283 (70 %) ze 406
nemocných dostávajících kladribin v dávce 5,25 mg/kg beze
známek aktivity onemocnění proti 145 pacientům (39 %)
z 373 ve skupině léčené placebem (OR 3,31; 95% CI
2,46‒4,46 pro skupinu s 3,5 mg/kg; OR 3,68;
95% CI 2,73‒4,97 pro skupinu s 5,25 mg/kg;
p < 0,0001).
V průběhu 48 týdnů bylo 208 (54 %) z 384 pacientů
ve skupině s kladribinem podávaným v dávce
3,5 mg/kg a 222 (56 %) z 396 pacientů ve skupině
s kladribinem v dávce 5,25 mg/kg beze známek aktivity
choroby proti 86 nemocným (24 %) ve skupině s placebem
(OR 3,80; 95% CI 2,77–5,22 pro kladribin v dávce 3,5 mg/kg;
OR 4,13; 95% CI 3,02‒5,66 pro kladribin v dávce 5,25 mg/kg;
p < 0,0001).
V průběhu 96 týdnů bylo 178 (44 %) ze 402 pacientů
ve skupině s kladribinem v dávce 3,5 mg/kg
a 189 (46 %) ze 411 pacientů ve skupině
s kladribinem v dávce 5,25 mg/kg bez aktivního
onemocnění proti 60 (16 %) z 379 pacientů ve sk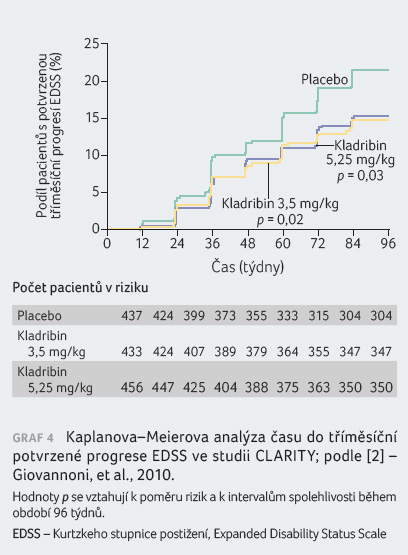 upině
s placebem (OR 4,28; 95% CI 3,05‒6,02 pro kladribin
v dávce 3,5 mg/kg; OR 4,62; 95% CI 3,29‒6,48 pro
kladribin v dávce 5,25 mg/kg; p < 0,0001).
Po dvou letech vykazovalo 47 % pacientů léčených
kladribinem (n = 391) stav NEDA (no evidence of disease
activity) stupně 3 proti 17 % léčených placebem (OR 4,25;
95% CI 3,03–6,96; p < 0,0001)
(obr. 3) [16].
upině
s placebem (OR 4,28; 95% CI 3,05‒6,02 pro kladribin
v dávce 3,5 mg/kg; OR 4,62; 95% CI 3,29‒6,48 pro
kladribin v dávce 5,25 mg/kg; p < 0,0001).
Po dvou letech vykazovalo 47 % pacientů léčených
kladribinem (n = 391) stav NEDA (no evidence of disease
activity) stupně 3 proti 17 % léčených placebem (OR 4,25;
95% CI 3,03–6,96; p < 0,0001)
(obr. 3) [16].
Post hoc analýza
byla provedena u pacientů s vysokou aktivitou onemocnění
léčených perorálním kladribinem v doporučené kumulativní
dávce 3,5 mg/kg. Patřili sem pacienti s jedním relapsem
v předchozím roce a s nejméně jednou Gd+ T1 lézí
nebo s devíti či více T2 lézemi během léčby jinými DMDs
nebo pacienti se dvěma nebo více relapsy v předchozím roce
léčení nebo neléčení pomocí DMDs. V podskupině nemocných
s vysokou aktivitou RS v porovnání s placebem
kladribin vykázal 67–68% redukci rizika relapsu, 82% redukci
rizika progrese disability, u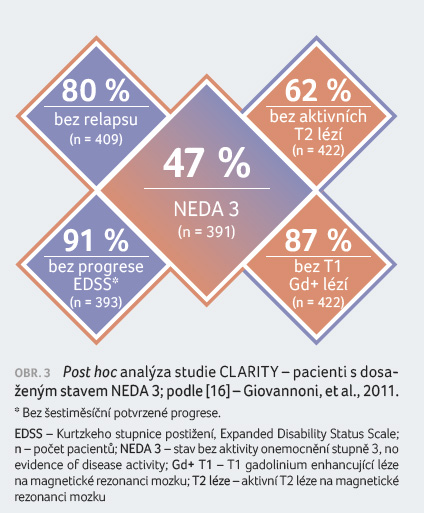 nemocných s EDSS vyšším
než 3,5 došlo k 47% redukci rizika relapsu [17].
nemocných s EDSS vyšším
než 3,5 došlo k 47% redukci rizika relapsu [17].
Pacienti, kteří dokončili studii CLARITY, mohli být zařazeni do její extenze (CLARITY EXT). V tomto následném 96týdenním období bylo 806 pacientů opět randomizováno, tentokrát do pěti skupin. Pacienti původně dostávající placebo byli dále léčeni kladribinem v dávce 3,5 mg/kg, nemocným původně léčeným kladribinem v dávce 3,5 mg/kg bylo dále podáváno placebo, nebo pokračovali třetím a čtvrtým rokem v pulzním podávání kladribinu (tedy v kumulativní dávce 7 mg/kg), a jedincům původně léčeným kladribinem v dávce 5,25 mg/kg bylo dále podáváno buď placebo, nebo kladribin v nižší dávce 3,5 mg/kg, tedy v kumulativní dávce 8,75 mg/kg. Klinické hodnocení zahrnovalo posouzení ARR a stupeň EDSS. Výchozí charakteristiky se mezi jednotlivými skupinami zásadně nelišily, i když pacienti dostávající placebo v původní studii CLARITY vykazovali vyšší klinickou a MR aktivitu. U skupin léčených původně kladribinem znamenalo pokračování v léčbě mírný přínos. Hodnota ARR u pacientů léčených kladribinem v dávce 3,5 mg/kg ve studii CLARITY a placebem ve studii CLARITY EXT byla 0,15 (97,5% CI 0,09‒0,21; n = 98), u pacientů, kteří pokračovali v léčbě kladribinem v dávce 3,5 mg/kg i třetím a čtvrtým rokem, byla hodnota ARR 0,10 (97,5% CI 0,06‒0,13; n = 186; p = 0,059). Obě skupiny vykazovaly srovnatelný podíl pacientů bez relapsu (75,6 % a 81,2 %) a srovnatelnou dobu manifestace dalšího relapsu. Střední hodnoty EDSS byly ve všech skupinách rovněž srovnatelné. Žádné významné rozdíly nebyly patrné ani v období vedoucím k potvrzení tříměsíční progrese EDSS. Studie CLARITY EXT ukázala, že klinický efekt kladribinu v dávce 3,5 mg/kg podávané v prvních dvou letech přetrvává nejméně po dobu čtyř let [18].
ORACLE byla dvojitě zaslepená, multicentrická, randomizovaná studie zkoumající účinek kladribinu na dobu konverze klinicky izolovaného syndromu (clinically isolated syndrome, CIS) do klinicky definitivní RS. Pacienti byli randomizováni k podávání kladribinu v dávce 3,5 mg/kg (n = 206), 5,25 mg/kg (n = 204) nebo placeba (n = 206) po dobu 96 týdnů. U pacientů léčených kladribinem bylo oproti placebu zřejmé snížení rizika přechodu do klinicky definitivní RS (HR [poměr rizik] 0,33 pro kladribin v dávce 3,5 mg/kg; 95% CI 0,21‒0,51; p < 0,0001; HR 0,38 pro kladribin v dávce 5,25 mg/kg; 95% CI 0,25‒0,58; p < 0,0001). Nežádoucí účinky byly hlášeny u 168 (82 %) pacientů ve skupině s kladribinem v dávce 3,5 mg/kg, 165 (81 %) pacientů ve skupině s kladribinem v dávce 5,25 mg/kg a 162 (79 %) pacientů ve skupině s placebem. Nebylo referováno žádné zvýšení rizika vzniku nežádoucích účinků aktivní léčby oproti placebu kromě lymfopenie, což byla závažná událost u čtyř (2 %) pacientů s dávkou kladribinu 3,5 mg/kg a u 10 (5 %) pacientů s dávkou kladribinu 5,25 mg/kg [3]. Vzhledem ke změně diagnostických kritérií (McDonald, 2010) byla provedena post hoc analýza pacientů retrospektivně splňujících či nesplňujících nová kritéria pro CIS. Kladribin v dávce 3,5 mg/kg (n = 68) snižoval riziko vzniku další klinické epizody nebo tříměsíční potvrzené progrese EDSS o 74 % proti placebu (n = 72; p = 0,0009). Kladribin v dávce 5,25 mg/kg snižoval riziko další epizody nebo zhoršení EDSS o 37 %, nebylo však dosaženo statistické významnosti [19].
ONWARD byla dvouletá randomizovaná, dvojitě zaslepená studie fáze IIb hodnotící účinek a bezpečnost kladribinu v dávce 3,5 mg/kg v kombinaci s interferonem beta 1b (IFNβ 1b) u pacientů s více než jedním relapsem během 48 týdnů při léčbě IFNβ 1b s EDSS 1,0–5,5. Průměrný počet relapsů byl nižší u pacientů léčených kladribinem v kombinaci s IFNβ 1b (n =124) ve srovnání s jedinci, kteří dostávali místo kladribinu placebo (0,56; n = 48). Hodnoty ARR představovaly 0,12 (95% CI 0,08‒0,17) a 0,32 (95% CI 0,20‒0,45). U pacientů léčených kladribinem bylo o 63 % méně pravděpodobné, že u nich dojde k relapsu (p < 0,001). Podíl pacientů s potvrzenou progresí EDSS se mezi oběma skupinami statisticky nelišil [20].
Zařazení do současné palety léčiv
Kladribin patří mezi léky ovlivňující průběh onemocnění u relabující remitující RS při selhání léčby první volby nebo u pacientů, u nichž od počátku probíhá onemocnění agresivně (více než jeden relaps ročně, aktivita na MR) a u nichž lze očekávat horší prognózu onemocnění. Doposud bylo možné tyto pacienty indikovat k podávání fingolimodu nebo dimetylfumarátu, eventuálně ‒ při výrazně vyšší aktivitě onemocnění ‒ k podávání natalizumabu nebo alemtuzumabu. Kladribin má podobné léčebné schéma jako alemtuzumab (dva léčebné pulzy v intervalu jednoho roku), jeho užívání je daleko komfortnější. Je možné jej podat i u žen v budoucnu plánujících graviditu bez nutnosti tzv. vymývací periody. Léčbu musí řídit a monitorovat lékař se zkušenostmi v terapii RS včetně imunosupresivních léčebných postupů.
Indikace a kontraindikace
Kladribin je indikován u dospělých pacientů s vysoce aktivní formou relabující remitující RS.
Mezi kontraindikace patří hypersenzitivita na léčivou látku nebo na kteroukoliv pomocnou látku, infekce virem lidské imunodeficience (HIV), aktivní chronická infekce (tuberkulóza nebo hepatitida), aktivní malignita, středně těžká nebo těžká porucha funkce ledvin (clearance kreatininu < 60 ml/min) a těhotenství nebo kojení. Zahájení léčby kladribinem není možné u imunokompromitovaných pacientů, včetně nemocných podstupujících v současné době imunosupresivní nebo myelosupresivní léčbu [20].
Nežádoucí účinky
Nežádoucí účinky referované ve studii CLARITY jsou
uvedeny v tabulce 2.
Problematika novotvarů je zmíněna v úvodu tohoto článku.
Z podrobnější analýzy bezpečnosti a tolerability
kladribinu ve studii CLARITY vyplývá, že lymfopenie byla
nejčastěji referovaným nežádoucím účinkem. Incidence infekcí
byla 48,3 % v případě kladribinu a 42,5 % v případě
placeba. Virus herpes zoster se manifestoval u 2,3 % pacientů
léčených kladribinem, žádný případ se nevyskytl ve skupině
s placebem. Zvláštní pozornost je nutno věnovat pacientům,
kteří se nesetkali s infekcí virem varicella zoster. Před
zahájením léčby kladribinem se doporučuje očkování pacientů
s negativitou protilátek a samotná terapie musí být
odložena o 4‒6 týdnů, aby byl umožněn plný účinek
očkování [21].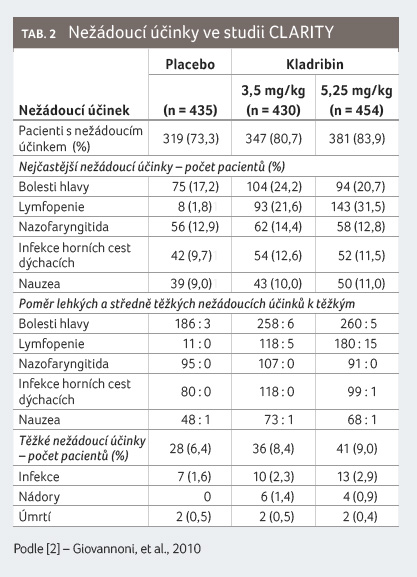
Z recentně publikovaných dat bezpečnostního profilu kladribinu v extenzi studie CLARITY vyplývá, že míra nežádoucích účinků byla mezi jednotlivými skupinami podobná, nicméně lymfopenie vyšší než stupně 3 se u jedinců léčených kladribinem v dávce 3,5 mg/kg vyskytovala častěji než u pacientů s placebem. U více než 90 % pacientů, u nichž se v období extenze objevila lymfopenie vyšší než stupně 3, došlo ke zlepšení tohoto onemocnění na stupeň 0–1 [22]. U nemocných s poklesem počtu lymfocytů pod 200 buněk/mm3 je doporučeno preventivní podávání acykloviru [21].
Podobné nálezy byly referovány v již zmíněné bezpečnostní integrované analýze. Virus herpes zoster byl hlášen častěji u pacientů, kteří vykazovali lymfopenii 3. nebo 4. stupně. U těchto pacientů je doporučeno preventivní podávání acykloviru. Souhrnná analýza dat z registru PREMIERE a z provedených klinických studií potvrzuje nízký výskyt nežádoucích účinků ‒ pouze u 3,5 % léčených vedly k přerušení terapie. V databázi klinických studií s kladribinem podávaným u RS (1 976 pacientů, 8 650 pacientoroků) nebyl hlášen žádný případ progresivní multifokální leukoencefalopatie (PML). Před zahájením léčby je však třeba provést výchozí vyšetření na MR (zpravidla během tří měsíců) [6].
Lékové interakce
Vzhledem k tomu, že kladribin obsahuje hydroxypropylbetadex, který může vést k tvorbě komplexů s dalšími léčivými přípravky (zejména s léčivými přípravky s nízkou rozpustností), může docházet ke zvýšení biologické dostupnosti takového přípravku. Proto se doporučuje, aby se jakýkoliv další perorální léčivý přípravek podával s odstupem alespoň tří hodin od užití kladribinu.
Zahájení léčby kladribinem je kontraindikováno u imunokompromitovaných pacientů, včetně pacientů, kteří v současné době podstupují imunosupresivní nebo myelosupresivní léčbu, např. metotrexátem, cyklofosfamidem, cyklosporinem nebo azathioprinem, nebo v případě chronického užívání kortikosteroidů z důvodu nebezpečí aditivních účinků na imunitní systém.
Krátkodobá akutní léčba systémovými kortikosteroidy, např. při léčbě relapsu, je během podávání kladribinu možná.
Současné podávání kladribinu s interferonem beta vede ke zvýšenému riziku lymfopenie. Bezpečnost a účinnost kladribinu v kombinaci s jinými typy léčby modifikujícími průběh choroby u RS nebyla stanovena. Souběžná léčba se nedoporučuje.
Z důvodu snížení počtu lymfocytů v důsledku léčby kladribinem lze očekávat výskyt aditivních hematologických nežádoucích účinků, jestliže se kladribin podává před užitím dalších přípravků, které ovlivňují hematologický profil (např. karbamazepin), nebo současně s nimi. V těchto případech se doporučuje důkladné sledování hematologických parametrů.
Na úrovni absorpce kladribinu se zdá být jediným možným mechanismem interakcí s klinickým významem protein rezistence karcinomu prsu (BCRP neboli ABCG2). Inhibice BCRP v gastrointestinálním traktu může zvýšit perorální biologickou dostupnost a systémovou expozici kladribinu. In vitro studie ukazují, že kladribin je substrátem ekvilibrativních nukleosidových (ENT1) a koncentrativních nukleosidových (CNT3) transportních proteinů. Biologická dostupnost, intracelulární distribuce a renální eliminace kladribinu může být následně ovlivněna silnými inhibitory transportérů ENT1 a CNT3, jako je například dilazep, nifedipin, nimodipin, cilostazol, sulindak nebo reserpin. Čisté účinky ve smyslu potenciálních změn expozice kladribinu je však těžké predikovat. Ačkoliv klinická závažnost těchto interakcí není známa, doporučuje se silné inhibitory ENT1, CNT3 nebo BCRP v době čtyř až pětidenní léčby kladribinem nepodávat současně. Není li to možné, je třeba zvážit výběr alternativních souběžně podávaných léčivých přípravků s žádnými nebo minimálními schopnostmi inhibice transportérů ENT1, CNT3 nebo BCRP. Pokud to nelze, doporučuje se upravit dávku léčivých přípravků obsahujících tyto látky na minimální nutné množství, přípravky podávat s časovým odstupem a pečlivě pacienta sledovat.
Účinky silných induktorů efluxních transportérů BCRP a P gp na biologickou dostupnost a expozici kladribinu nebyly formálně hodnoceny. Mělo by být zváženo možné snížení expozice kladribinu při současném podávání silných induktorů transportérů BCRP (např. kortikosteroidy) nebo P gp (např. rifampicin, třezalka tečkovaná).
V současné době není známo, zda může kladribin snižovat účinnost systémově působící hormonální antikoncepce. U žen, které používají systémově působící hormonální antikoncepci, je tedy nutné během léčby kladribinem a po dobu nejméně čtyř týdnů po poslední dávce v každém léčebném roce přidání bariérové metody antikoncepce [21].
Zvláštní populace
U pacientů s poruchou funkce ledvin nebyly provedeny žádné specifické studie. U pacientů s lehkou poruchou funkce ledvin (clearance kreatininu 60‒89 ml/min) není nutná žádná úprava dávkování. Bezpečnost a účinnost u pacientů se středně těžkou nebo těžkou poruchou funkce ledvin nebyla stanovena. Z tohoto důvodu je kladribin u těchto nemocných kontraindikován. U pacientů s poruchou funkce jater nebyly provedeny žádné studie. Ačkoliv význam funkce jater pro eliminaci kladribinu je považován za zanedbatelný, není vzhledem k chybějícím údajům užívání kladribinu doporučeno u nemocných se středně těžkou nebo těžkou poruchou funkce jater (Childovo‒Pughovo skóre > 6).
Klinické studie s perorálním kladribinem u RS nezahrnovaly pacienty starší 65 let, proto není známo, zda reagují na léčbu jinak než mladší nemocní. Bezpečnost a účinnost kladribinu u pacientů ve věku do 18 let nebyla stanovena. Nejsou dostupné žádné údaje [21].
Těhotenství a kojení
U žen ve fertilním věku musí být před zahájením léčby kladribinem v prvním a v druhém roce vyloučeno těhotenství a musí mu být zabráněno použitím účinné antikoncepce během terapie a po dobu nejméně šesti měsíců po poslední dávce přípravku. U žen, které používají systémově působící hormonální antikoncepci, je nutné během léčby kladribinem a po dobu nejméně čtyř týdnů po poslední dávce v každém léčebném roce přidání bariérové metody antikoncepce. Ženy, které otěhotní během léčby kladribinem, jsou nuceny terapii ukončit.
Jelikož kladribin ovlivňuje syntézu DNA, je možné očekávat nežádoucí účinky na gametogenezi u člověka. Pacienti mužského pohlaví musejí proto učinit opatření, aby zajistili, že jejich partnerky během terapie kladribinem a nejméně šest měsíců po poslední dávce neotěhotní. Zkušenosti získané u člověka s jinými látkami inhibujícími syntézu DNA naznačují, že kladribin podávaný během těhotenství by mohl způsobit vrozené vady. Studie na zvířatech prokázaly reprodukční toxicitu.
Není známo, zda se kladribin vylučuje do lidského mateřského mléka. Vzhledem k možným závažným nežádoucím účinkům u kojených dětí je kojení během léčby kladribinem a jeden týden po poslední dávce přípravku kontraindikováno [21].
Dávkování
Doporučená kumulativní dávka kladribinu je 3,5 mg/kg tělesné
hmotnosti v průběhu dvou let podávaná jako jeden léčebný
pulz v dávce 1,75 mg/kg za rok. Každý léčebný pulz
zahrnuje dva týdny léčby, jeden na začátku prvního m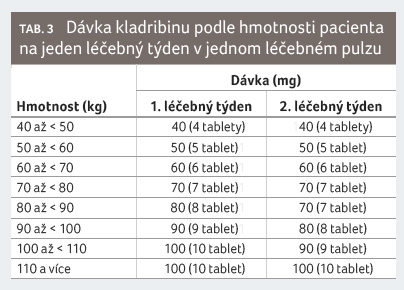 ěsíce
a jeden na začátku druhého měsíce příslušného
léčebného roku. Každý léčebný týden zahrnuje čtyři nebo
pět dnů, během nichž dostane pacient 10 mg nebo 20 mg
kladribinu (jednu nebo dvě tablety) v jedné denní dávce
v závislosti na tělesné hmotnosti. Vynechaná dávka se
nesmí užít společně s další plánovanou dávkou
následující den. Pokud dojde k vynechání dávky, musí
pacient vynechanou dávku užít následující den a počet dnů
v léčebném týdnu se zvýší. Pokud pacient zapomene užít
dvě po sobě následující dávky, platí stejné pravidlo
a počet dnů v léčebném týdnu se prodlouží o dva
dny. Po dokončení dvou léčebných pulzů není během
třetího a čtvrtého roku nutná žádná další léčba.
Opakované zahájení léčby po čtvrtém roce nebylo
hodnoceno. Před zahájením prvního pulzu musí být počet
lymfocytů v normě. Před zahájením druhého pulzu (v druhém
roce) musí být před zahájením léčby počet lymfocytů
minimálně 800 buněk/mm3. Je li to nutné, může
se léčebný pulz v druhém roce odložit až o šest
měsíců, než se koncentrace lymfocytů upraví. Jestliže je čas
nutný k úpravě delší než šest měsíců, nemá se léčba
opakovat. Dávkovací schéma je patrné z tabulky 3 a 4
[21].
ěsíce
a jeden na začátku druhého měsíce příslušného
léčebného roku. Každý léčebný týden zahrnuje čtyři nebo
pět dnů, během nichž dostane pacient 10 mg nebo 20 mg
kladribinu (jednu nebo dvě tablety) v jedné denní dávce
v závislosti na tělesné hmotnosti. Vynechaná dávka se
nesmí užít společně s další plánovanou dávkou
následující den. Pokud dojde k vynechání dávky, musí
pacient vynechanou dávku užít následující den a počet dnů
v léčebném týdnu se zvýší. Pokud pacient zapomene užít
dvě po sobě následující dávky, platí stejné pravidlo
a počet dnů v léčebném týdnu se prodlouží o dva
dny. Po dokončení dvou léčebných pulzů není během
třetího a čtvrtého roku nutná žádná další léčba.
Opakované zahájení léčby po čtvrtém roce nebylo
hodnoceno. Před zahájením prvního pulzu musí být počet
lymfocytů v normě. Před zahájením druhého pulzu (v druhém
roce) musí být před zahájením léčby počet lymfocytů
minimálně 800 buněk/mm3. Je li to nutné, může
se léčebný pulz v druhém roce odložit až o šest
měsíců, než se koncentrace lymfocytů upraví. Jestliže je čas
nutný k úpravě delší než šest měsíců, nemá se léčba
opakovat. Dávkovací schéma je patrné z tabulky 3 a 4
[21].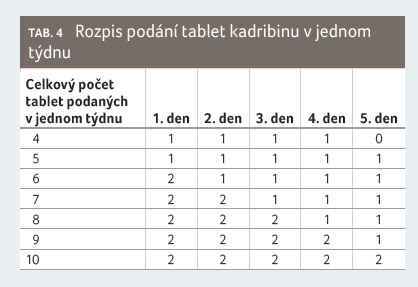
Závěr
Kladribin představuje novou šanci pro pacienty s RR RS. Kromě významné redukce ARR, snížení podílu nemocných bez relapsu po dobu 96 týdnů, podílu nemocných bez trvalé invalidity po dobu 96 týdnů a doby do tříměsíční progrese EDSS v porovnání s pacienty v placebové skupině, redukce mozkové atrofie a snížení počtu aktivních lézí na MR byl prokázán i významný efekt u nemocných s vysokou aktivitou onemocnění. Léčivo má uspokojivý bezpečnostní profil a komfortní dávkování s podáním v léčebných pulzech ve dvou následných letech. Počet tablet závisí na hmotnosti pacienta, kdy celková dávka ve dvou letech léčby je 3,5 mg na kilogram tělesné hmotnosti. Lze očekávat, že kladribin si najde velmi brzy své pevné místo ve stále se rozšiřující paletě léčebných možností u RS.
Seznam použité literatury
- [1] Beutler E, Sipe JC, Romine JS, et al. The treatment of chronic progressive multiple sclerosis with cladribine. Proc Natl Acad Sci U S A 1996; 93: 1716‒1720.
- [2] Giovannoni G, Comi G, Cook S, et al. CLARITY Study Group. A placebo‑controlled trial of oral cladribine for relapsing multiple sclerosis. N Engl J Med 2010; 362: 416‒426.
- [3] Leist TP, Comi G, Cree BA, et al. Oral cladribine for early MS (ORACLE MS) Study Group. Effect of oral cladribine on time to conversion to clinically definite multiple sclerosis in patients with a first demyelinating event (ORACLE MS): a phase 3 randomised trial. Lancet Neurol 2014; 13: 257‒267.
- [4] Kappos L, Radue EW, O’Connor P, et al. FREEDOMS Study Group. A placebo‑controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med 2010; 362: 387‒401.
- [5] Pakpoor J, Disanto G, Altmann DR, et al. No evidence for higher risk of cancer in patients with multiple sclerosis taking cladribine. Neurol Neuroimmunol Neuroinflamm 2015; 2: e158.
- [6] Cook S, Leist T, Comi G, et al. Cladribine Tablets in the Treatment of Patients with Multiple Sclerosis: An Integrated Analysis of Safety from the Multiple Sclerosis Clinical Development Program. Neurology 2017; 88(Suppl 16): Abstract P5.394.
- [7] Sigal DS, Miller HJ, Schram ED, et al. Beyond hairy cell: the activity of cladribine in other hematologic malignancies. Blood 2010; 116: 2884‒2896.
- [8] Schreiber K, Soelberg Sorensen P. Cladribine in the treatment of multiple sclerosis. Clin Invest 2011; 1: 317–326.
- [9] Mitosek‑Szewczyk K, Tabarkiewicz J, Wilczynska, et al. Impact of cladribine therapy on changes in circulating dendritic cell subsets, T cells and B cells in patients with multiple sclerosis. J Neurol Sci 2013; 332: 35‒40.
- [10] Giovannoni G. Cladribine to Treat Relapsing Forms of Multiple Sclerosis. Neurotherapeutics 2017; 14: 874‒887.
- [11] Wiendl H. Cladribine ‒ an old newcomer for pulsed immune reconstitution in MS. Nat Rev Neurol 2017; 13: 573‒574.
- [12] Lindemalm S, Savic RM, Karlsson MO, et al. Application of population pharmacokinetics to cladribine. BMC Pharmacol 2005; 5: 4.
- [13] Savic RM, Novakovic AM, Ekblom M, et al. Population Pharmacokinetics of Cladribine in Patients with Multiple Sclerosis. Clin Pharmacokinet 2017; 56: 1245‒1253.
- [14] De Stefano N, Giorgio A, Battaglini M, et al. Reduced brain atrophy rates are associated with lower risk of disability progression in patients with relapsing multiple sclerosis treated with cladribine tablets. Mult Scler 2017; doi: 10.1177/1352458517690269 [Epub ahead of print]
- [15] Cook A, Rammohan K, Rieckmann P, et al. Slowing of Disability Progression Based on 6‑Month Confirmed EDSS in Patients with Relapsing‑Remitting Multiple Sclerosis (RRMS) Treated with Cladribine Tablets in the CLARITY Study: A Post‑Hoc Subgroup Analysis. Neurology 2016; 86(Suppl 16): Abstract P3.058.
- [16] Giovannoni G, Cook S, Rammohan K, et al. CLARITY study group. Sustained disease‑activity‑free status in patients with relapsing‑remitting multiple sclerosis treated with cladribine tablets in the CLARITY study: a post‑hoc and subgroup analysis. Lancet Neurol 2011; 10: 329‒337.
- [17] Giovannoni G, Rammohan K, Cook S, et al. Efficacy of Cladribine Tablets 3.5 mg/kg in High Disease Activity (HDA) Subgroups of Patients with Relapsing Multiple Sclerosis (RMS) in the CLARITY Study. Neurology 2017; 88(Suppl 16): Abstract P6.360.
- [18] Giovannoni G, Comi G, Cook S, et al. Clinical Efficacy of Cladribine Tablets in Patients with Relapsing‑Remitting Multiple Sclerosis (RRMS): Final Results from the 120‑Week Phase IIIb Extension Trial to the CLARITY Study. Neurology 2016; 86(Suppl 16): Abstract P3.028.
- [19] Freedman MS, Leist TP, Comi G, et al. The efficacy of cladribine tablets in CIS patients retrospectively assigned the diagnosis of MS using modern criteria: Results from the ORACLE MS study. Mult Scler J Exp Transl Clin 2017; 3: 234‒240.
- [20] Montalban X, Cohen B, Leist T, et al. Efficacy of Cladribine Tablets as Add‑On to IFN‑beta Therapy in Patients with Active Relapsing MS: Final Results from the Phase II ONWARD Study (P3.029). AAN 2016. Dostupné na: http://www.abstractsonline.com/pp8/#!/4046/presentation/9728
- [21] Mavenclad (kladribin). Souhrn údajů o přípravku. Státní ústav pro kontrolu léčiv. Dostupné na: http://www.ema.europa.eu/docs/cs_CZ/document_library/EPAR_Product_Information/human/004230/WC500234561.pdf [navštíveno 8. 1. 2018]
- [22] Cook S, Vermersch P, Comi G, et al. CLARITY Study Group. Safety and tolerability of cladribine tablets in multiple sclerosis: the CLARITY (CLAdRIbine Tablets treating multiple sclerosis orallY) study. Mult Scler 2011; 17: 578‒593.
- [23] Giovannoni G, Soelberg Sorensen P, Cook S et al. Safety and efficacy of cladribine tablets in patients with relapsing‑remitting multiple sclerosis: Results from the randomized extension trial of the CLARITY study. Mult Scler 2017; doi: 10.1177/1352458517727603. [Epub ahead of print]