Nelarabin
Léčba relabovaných nebo rezistentních případů akutní lymfoblastické leukemie (ALL) nebo lymfoblastického lymfomu (LBL) je velmi svízelná. Jen menšinu pacientů s relabovanou ALL nebo relabovaným LBL se podaří stávajícími léčebnými postupy zachránit. Nelarabin, nové léčivo ze skupiny nukleosidových analogů, může už v monoterapii navodit významné procento kompletních remisí u rezistentních ALL nebo LBL z T-lymfocytární řady. Remise ale většinou nejsou trvalé, a proto by měla na léčbu nelarabinem navazovat alogenní transplantace krvetvorby. V takovýchto případech představuje lék novou naději pro nemocné s relabovanými nebo rezistentními T-ALL a T-LBL.
Akutní lymfoblastická leukemie (ALL) je onemocnění, jehož incidence má dvojí vrchol. První se objevuje kolem 4. roku života, druhý pak ve věku nad 60 let [1]. Už od roku 1975 je známo, že většinu případů (kolem 60–80 %) tohoto onemocnění tvoří ALL, které vycházejí z B lymfocytů. Zbytek pak tvoří ALL z T buněk [2, 3]. U dětí se v současné době daří vyléčit až 90 % nemocných s ALL. Většinou u nich není třeba ani použít transplantaci krvetvorných buněk. U dospělých je situace mnohem horší. Dlouhodobě přežívá podle různých zpráv 30–50 % dospělých nemocných s ALL. Procento vyléčených se u dospělých, na rozdíl od dětí, od 90. let 20. století příliš nemění, a to i přes zlepšující se podpůrnou terapii, hlubší poznání biologie ALL a nové, často individualizované léčebné režimy [4–6]. Jednou z příčin této situace je fakt, že u dětí nacházíme více prognosticky příznivých cytogenetických a molekulárně-genetických změn v blastech ALL ve srovnání s dospělými, další příčinou je skutečnost, že dospělí hůře tolerují intenzivní léčbu. V neposlední řadě stojí za špatnými výsledky léčby ALL dospělých také zjištění, že jejich ALL blasty jsou rezistentnější na léčiva, která se v terapii ALL používají. Blasty dospělých s ALL jsou 31,7krát rezistentnější k prednisonu, 6,9krát k dexamethasonu, 6,1krát k L-asparagináze, 2,9krát k cytarabinu, 2,5krát k daunorubicinu a 2,2krát k vinkristinu ve srovnání s blasty dětských ALL [7]. Jak naznačují předešlé údaje, velkým problémem u pacientů s ALL, zejména dospělých pacientů s ALL, jsou relapsy onemocnění. Relapsy ALL představují jednu z nejméně radostných oblastí současné hematologie. Je stále otázkou, jak v případě ralapsu ALL postupovat. Podle analýzy výsledků léčby relabované T-ALL a jí příbuzného T lymfoblastického lymfomu (LBL) podle německého protokolu GMALL 05-1993 je kompletní remise (CR) dosaženo jen u 29 % pacientů s relabovanými T-ALL nebo T-LBL a dlouhodobě přežívá jen 8 % těchto nemocných [8, 9]. Jednou z nadějí pro pacienty s relabovanou ALL z T řady se v současné době stává nelarabin.
Farmakologická skupina
Nelarabin (506U78) je cytostatikum ze skupiny nukleosidových analogů [10], ATC kód: L01BB07.
Chemické a fyzikální vlastnosti
Chemický název nelarabinu je 2-amino-9-b-D-arabinofuranosyl-6-methoxy-9H-purin (obr. 1).
Sumární vzorec: C11H15N5O5
Molekulová hmotnost: 297,27
Nelarabin je dobře rozpustný ve vodě (na rozdíl od 9-b-D-arabinofuranosylguaninu, viz níže). Rozkládá se při teplotách mezi 209–217 °C [11, 12].
Mechanismus účinku, farmakodynamika
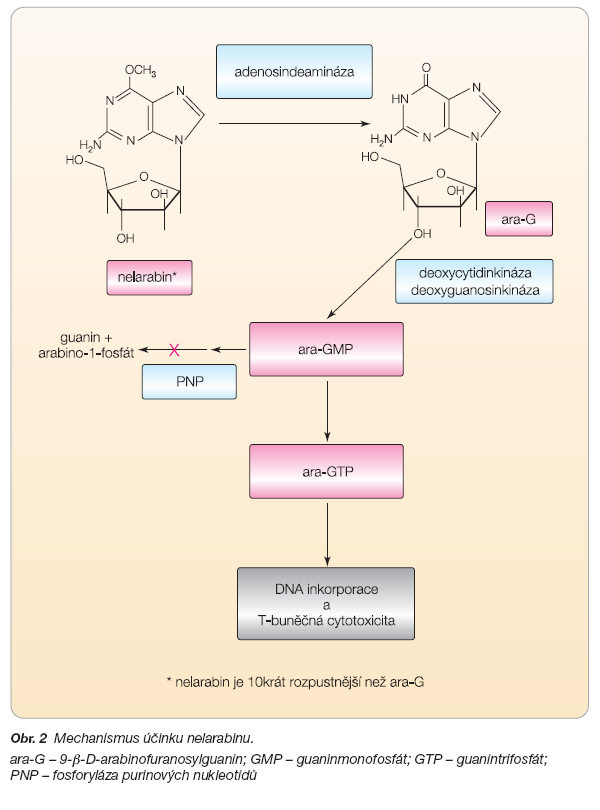
Nelarabin je prodrug cytotoxického deoxyguanosinového analogu 9-b-D-arabinofuranosylguaninu (ara-G). Nelarabin je po aplikaci demetylován adenosindeaminázou (ADA) na ara-G. Dále je fosforylován deoxyguanosinkinázou a deoxycytidinkinázou na aktivní trifosfát ara-GTP. Akumulace ara-GTP v blastech ALL a jeho inkorporace do jejich deoxyribonukleové kyseliny (DNA) vede k inhibici syntézy DNA a k buněčné smrti [11, 12], viz obr. 2.
Nelarabin je vysoce účinný v inhibici růstu T lymfoblastů. Ve srovnání s dalším novým léčivem pro léčbu ALL klofarabinem je in vitro u T-ALL 188krát účinnější. Naopak na B lymfoblasty působí klofarabin 7krát silněji než nelarabin [13].
Ara-G poprvé syntetizovali už v 50. letech 20. století Gertruda Elionová a George Hitchings, kteří pracovali na výzkumu nových imunosupresiv. Ara-G byl syntetizován jako inhibitor fosforylázy purinových nukleotidů (PNP), o níž bylo známo, že její afunkčnost způsobuje těžký deficit T lymfocytů [14]. Imunosupresivní aktivita ara-G byla ale nízká, a tak na 20 let upadl do zapomnění. Až v roce 1983 začala být zkoumána jeho aktivita proti buňkám lidských leukemií [15]. Následně byl syntetizován prodrug ara-G, který dostal označení 506U78 (nelarabin) a jenž byl mnohem více solubilní než ara-G [16].
Farmakokinetickévlastnosti
Základní farmakokinetické parametry nelarabinu a ara-G u dospělých a dětí shrnuje tab. 1.
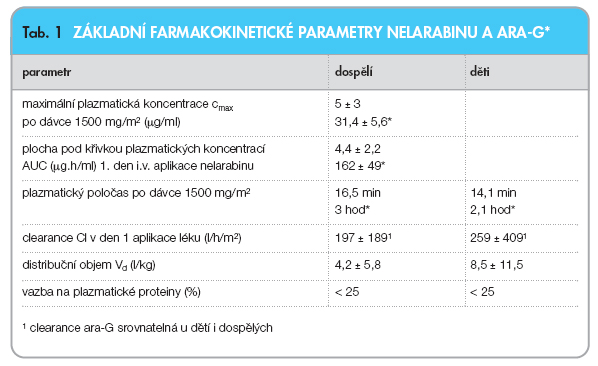
Farmakokinetické studie u dospělých pacientů s refrakterními leukemiemi a lymfomy ukázaly, že nelarabin (podaný v dávce 1500 mg/m2) je z plazmy dospělých eliminován za 16,5 minuty a ara-G za 3 hodiny. Poločas nelarabinu u dětí je 14,1 minuty, poločas ara-G je u nich 2,1 hodiny. Plazmatická hladina cmax je u ara-G nejvyšší ke konci infuze nelarabinu. Cmax ara-G je vyšší než cmax nelarabinu, což prokazuje velmi rychlou přeměnu nelarabinu na ara-G. Po podání nelarabinu v dávce 1500 mg/m2 (infuze 2 hodiny) je průměrná cmax nelarabinu 5 Î 3 mg/ml a průměrná cmax ara-G 31,4 Î 5,6 mg/ml. Plocha pod křivkou (AUC) ara-G je v první den intravenózní aplikace nelarabinu (1500 mg/m2) 37krát větší než AUC nelarabinu (162 Î 49 mg.h/ml versus 4,4 Î 2,2 mg.h/ml). Cmax a AUC nelarabinu jsou v prvních 5 dnech aplikace léku srovnatelné; to nasvědčuje tomu, že farmakokinetika nelarabinu podávaného v několika dávkách je odvoditelná z dávky jediné. Pro ara-G nejsou přesná data o cmax a AUC v prvních 5 dnech aplikace nelarabinu známa. Po první infuzi nelarabinu v dávce 1500 mg/m2 je cmax ara-GTP dosaženo intracelulárně během 3. až 25. hodiny po aplikaci. AUC intracelulárního ara-GTP je 532krát vyšší než AUC intracelulárního nelarabinu a 14krát vyšší než AUC intracelulárního ara-G. Jelikož intracelulární přítomnost ara-GTP je velmi prolongovaná, poločas jeho eliminace nebylo možné přesně stanovit.
Farmakokinetická studie fáze 1 u nelarabinu ukázala, že jeho clearance je u dětí v den 1 aplikace léku asi o 30 % vyšší než u dospělých (259 Î 409 l/h/m2 versus 197 Î 189 l/h/m2), zatímco clearance ara-G je u dětí i dospělých srovnatelná. Nelarabin a ara-G jsou distribuovány po celém organismu. Distribuční objem nelarabinu je u dospělých 4,2 Î 5,8 l/kg, u dětí 8,5 Î 11,5 l/kg. Nelarabin ani ara-G se in vitro významně nevážou na lidské plazmatické proteiny (< 25 %). Tato vazba je nezávislá na koncentracích nelarabinu a ara-G, pokud jsou vyšší než 600 mM.
Nelarabin je metabolizován O-demetylací zprostředkovanou ADA. Vzniklé ara-G je hydrolyzováno na guanin, případně metylguanin. Guanin je přeměněn na xantin, jenž se oxiduje na kyselinu močovou a ta se mění na alantoin.
Nelarabin a ara-G jsou vylučovány ledvinami. Průměrná renální exkrece nelarabinu a ara-G je za 24 hodin 6,6 Î 4,7 %, resp. 27 Î 15 % z celkové podané dávky v den 1. Studie fáze 1 ukázala, že clearance nelarabinu a jeho distribuční objem jsou vyšší cca o 10 % u bělochů ve srovnání s černochy. Naopak pro ara-G jsou tyto parametry vyšší o 15–20 % u černochů. V účinnosti a bezpečnosti léčiva nebyly pozorovány žádné rasové rozdíly. Farmakokinetika nelarabinu a farmakokinetika ara-G nejsou ovlivněny ani věkem, nicméně horší renální funkce u starších lidí mohou snížit clearance ara-G. Mírně až středně zhoršené renální parametry (kreatininová clearance mezi 50–80 ml/min) vedou u nemocných ke snížení clearance ara-G o 15–40 %. Vliv zhoršených funkcí jater na farmakokinetiku nelarabinu studován nebyl [11, 12, 17, 18].
Klinické zkušenosti
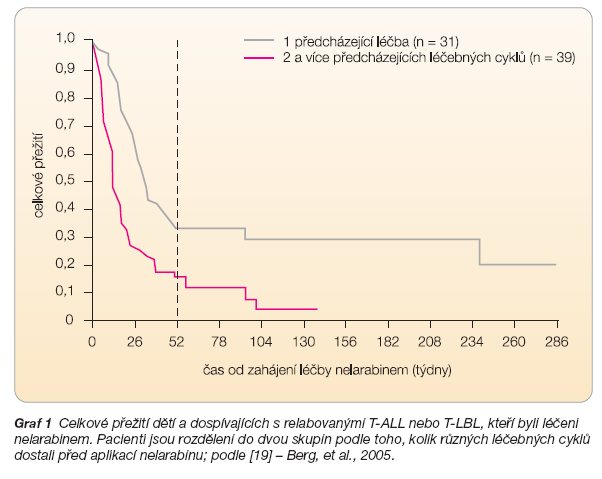
S nelarabinem bylo provedeno několik klinických studií. První z nich byla zahájena v roce 1993. Bezpečnost a účinnost nelarabinu u dětských pacientů a adolescentů s relabovanými a rezistentními T-ALL a T-LBL zkoumala studie Children s Oncology Group (COG P9673) [19]. Tato studie s dávkou nelarabinu 650 mg/m2 intravenózně den 1–5 byla provedena u 63 nemocných s mediánem věku 11,9 let (2,5–21,7). Třicet z nich bylo předléčeno dvěma a více jinými léčebnými režimy, zbytek nemocných jedním režimem. Příznivé léčebné odpovědi bylo dosaženo u 55 % nemocných v prvním relapsu a u 27 % nemocných ve druhém a dalším relapsu T-ALL či T-LBL. Ze všech nemocných léčených výše uvedenou dávkou dosáhlo 38 % nemocných kompletní remise nemoci (CR) a 5 % parciální remise (PR). Trvání CR bylo 3,3–9,3 týdne. Medián celkového přežití (OS) byl 13,1 týdne, viz graf 1. V této studii bylo hodnoceno ještě dalších 73 nemocných, kteří dostávali nelarabin v jiné dávce (400 nebo 900 mg/m2 den 1–5) nebo měli relaps ALL v centrálním nervovém systému. Výsledky léčby těchto nemocných byly horší než u pacientů, kteří dostávali dávku 650 mg/m2 den 1–5 [19].
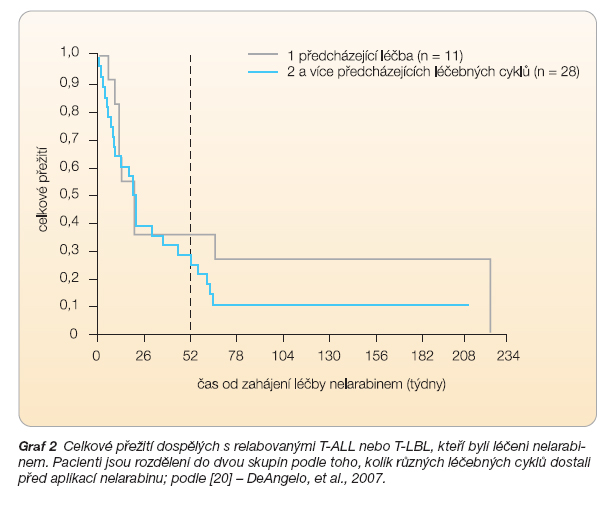
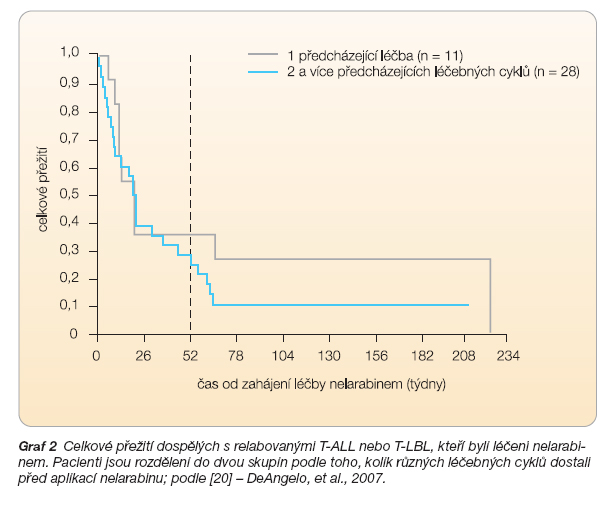
Ve studii Cancer and Leukemia Group B (CALGB 19801) bylo léčeno 39 dospělých nemocných s rezistentními nebo relabovanými T-ALL a T-LBL. Celkem 28 z těchto nemocných bylo před podáním nelarabinu léčeno nejméně dvěma různými terapeutickými režimy. Nelarabin byl nemocným podán v dávce 1500 mg/m2 intravenózně v den 1, 3 a 5. Infuze trvala 2 hodiny. Cyklus léčby se opakoval po 21 dnech. CR nebo PR dosáhlo 41 % nemocných. CR bylo dosaženo u 26 % nemocných, CR* (CR bez kompletní hematologické obnovy) u 5 % pacientů. Medián OS byl 20,6 týdne [20], viz graf 2.
Studie německých autorů s nelarabinem u relabovaných T-ALL a T-LBL, v níž bylo hodnoceno 53 dospělých nemocných, prokázala vysokou účinnost nelarabinu v této skupině pacientů. Po podání léčiva bylo dosaženo 47 % CR a 13 % PR. U 19 nemocných bylo možné následně provést alogenní transplantaci krvetvorby. Z pacientů, kteří po podání nelarabinu dosáhli CR a byli následně transplantováni, dlouhodobě přežívalo 27 % osob [9]. Goy a kol. prezentovali už v roce 2003 na mítinku Americké hematologické společnosti 18 % CR a 36 % PR u pacientů s ne-Hodgkinovými lymfomy léčenými nelarabinem [21]. Kurtzbergová a kol. dosáhli u heterogenní skupiny 93 pacientů s refrakterními lymfoidními malignitami 11 % CR a 20 % PR po podání nelarabinu. V podskupině pacientů s T-ALL (n = 39) bylo dosaženo 23 % CR a 31 % PR [22]. Czuczman a kol. popsali účinnost nelarabinu u pacientů s rezistentními a relabovanými periferními nebo kožními T lymfomy. U celkem 19 nemocných bylo dosaženo jen 2 PR a žádné CR. Jedna PR trvala 3 měsíce a druhá 5,5 měsíce [23]. Souhrn hlavních klinických studií s nelarabinem ukazuje tab. 2.
Zařazení do současné palety léčiv
Nelarabin rozšiřuje spektrum léčebných možností pro pacienty s T-ALL a T-LBL. Zatím je indikován jen v monoterapii. Lze předpokládat, že v budoucnu bude kombinován s dalšími protinádorovými léčivy účinnými u těchto nemocných.
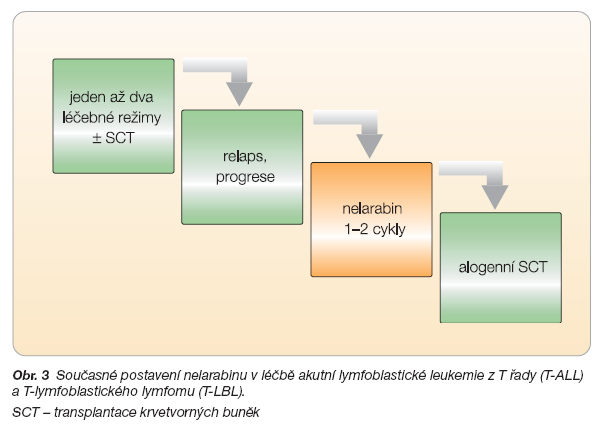
Současné postavení nelarabinu v léčbě T-ALL a T-LBL shrnuje obr. 3.
Indikace
Nelarabin je indikován u pacientů s relabovanou nebo na léčbu první a druhé linie rezistentní ALL z T řady nebo u pacientů s relabovaným nebo rezistentním T-LBL. Měl by být podáván zejména pacientům, u kterých je naděje, že budou moci následně podstoupit alogenní transplantaci krvetvorby [11, 12].
Kontraindikace
Nelarabin je kontraindikován u pacientů s prokázanou přecitlivělostí na lék. Dále by lék neměl být indikován v graviditě. U březích králíků jeho podání zvyšuje incidenci malformací plodu (rozštěpy patra, chybění prstů, chybění žlučníku, snížení počtu nebo nadbytek plicních laloků, splynutí obratlů, opožděná osifikace skeletu) a snižuje hmotnost plodu. Přípravek nesmí být podáván kojícím matkám, resp. všechny nemocné, které kojí, by měly kojení ukončit [11, 12].
Nežádoucí účinky
Podání nelarabinu může být spojeno s významnými nežádoucími účinky. Nejvýznamnějším toxickým projevem nelarabinu je neurotoxicita, která limituje podanou dávku léčiva. K symptomům nelarabinem indukované neurotoxicity patří somnolence (nejčastěji přímo v den aplikace), zmatenost, křeče, ataxie, parestezie a hypoestezie. Parestezie a hypoestezie mohou přetrvávat měsíce po podání léku. Závažná neurologická toxicita může vyústit až v kóma, status epilepticus, kraniospinální demyelinizaci nebo vzestupnou neuropatii, která se podobá syndromu Guillain-Barré. Zvýšené riziko neurologické toxicity se týká především nemocných, kteří jsou zároveň s nelarabinem léčeni intratekální aplikací chemoterapie. Riziko toxicity léčiva roste s jeho dávkou. Při projevech neurotoxicity stupně 2 a vyšší by mělo být podávání léku ukončeno. Incidenci neurotoxicity přibližují tab. 3 a tab. 4.
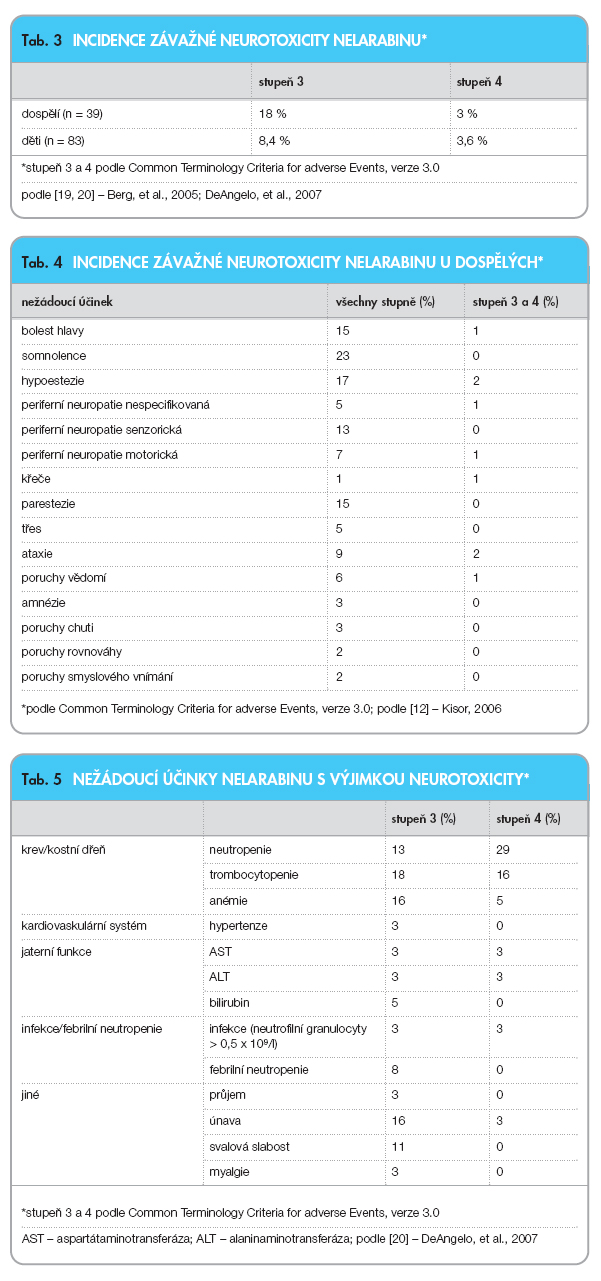
K hematologickým nežádoucím účinkům nelarabinu patří leukopenie, neutropenie, trombocytopenie a anémie. K dalším nežádoucím účinkům nelarabinu paří průjem, myalgie, hypokalémie, hypalbuminémie, hyperbilirubinémie či elevace jaterních transamináz. Zvracení stupně 3 nebo 4 (podle Common Terminology Criteria for adverse Events, verze 3.0) léčivo vyvolává vzácně [11, 19, 20]. Nežádoucí účinky nelarabinu shrnuje tab. 5.
Vzhledem k vyššímu riziku toxicity nelarabinu u pacientů se zhoršenou funkcí ledvin a jater by měli být nemocní s kreatininovou clearance nižší než 30 ml/min nebo s plazmatickou hladinou bilirubinu vyšší než 3 mg/dl intenzivněji sledováni stran nežádoucích účinků léčiva. Stejně tak nemocní starší 65 let by měli být důkladněji sledováni, neboť u nich je také riziko toxicity nelarabinu (zejména neurologické) zvýšeno [11, 12].
Lékové interkace
Nelarabin ani ara-G in vitro významně neinhibují nebo neaktivují izoenzymy lidského jaterního cytochromu P-450 (1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6 nebo 3A4). U pacientů s refrakterními leukemiemi nebyla farmakokinetika nelarabinu, ara-G ani ara-GTP ovlivněna 30minutovou infuzí fludarabinu v dávce 30 mg/m2, která infuzi nelarabinu předcházela o 4 hodiny [11, 12].
Dávkování
Z klinických studií CALGB19801 a PGAA2003 (celkem 103 pacientů) vyplynula dávka léčiva pro dospělé: 1500 mg/m2 ve dny 1, 3, 5 intravenózně. Jednotlivá infuze by měla být aplikována 2 hodiny. Tuto kúru lze opakovat za 21 dní. Při projevech neurotoxicity léčiva stupně 2 a vyšší by mělo být podávání léku ukončeno. Ze studie COG P9673 (84 pacientů) vzešla dávka nelarabinu pro děti: 650 mg/m2 denně po dobu 5 dní. Proti předávkování lékem neexistuje antidotum [11, 19, 20].
Balení
Nelarabin je dodáván jako čirý a bezbarvý roztok ve skleněných ampulích. Každá 50ml ampule obsahuje aqua pro injectione, v níž je rozpuštěno 250 mg nelarabinu (5 mg nelarabinu v 1 ml) a chlorid sodný (4,5 mg v 1 ml); pH roztoku se pohybuje mezi 5–7 [11, 12]. Ve Spojených státech amerických je nelarabin registrován pod názvem Arranon, v Evropě pod názvem Atriance, viz tab. 6. V Evropské unii je podávání nelarabinu schváleno od srpna 2007.