Nivolumab
Souhrn:
Fínek J. Nivolumab. Remedia 2019; 29: 241–246.
Nivolumab je plně humánní protilátka IgG4, která selektivně blokuje inhibiční receptor programované buněčné smrti 1 (PD‑1) exprimovaný na aktivovaných cytotoxických T lymfocytech a umožňuje restart protinádorové odpovědi. Léčivá látka je aktivní v terapii řady malignit.
Summary:
Finek J. Nivolumab. Remedia 2019; 29: 241–246.
Nivolumab is a fully human IgG4 antibody that selectively blocks programmed cell death inhibiting receptor 1 (PD‑1) expressed on activated cytotoxic T‑lymphocytes and allows restarting antitumor response. Nivolumab is active in the therapy of many malignancies.
Key words: immunotherapy, antibody, carcinoma.
Úvod
Nivolumab (MDX 1106, BMS 936558) je plně humánní protilátka IgG4, která selektivně blokuje inhibiční receptor programované buněčné smrti 1 (programmed cell death protein 1, PD 1) exprimovaný na aktivovaných cytotoxických T lymfocytech. Blokuje tím vazbu na ligandy PD 1/PD 2 (PD L1/PD L2), které exprimují nádorové buňky, a umožní restart imunitní protinádorové reakce. Obnovená aktivita vlastního imunitního systému pak dokáže navodit objektivní protinádorovou odpověď, stabilizovat onemocnění, a v některých případech dokonce nemocného s metastatickým onemocněním vyléčit. Jako jeden z dostupných imunoterapeutických léků prokázal nivolumab svou účinnost u řady nádorových onemocnění.
V současné době je PD 1 checkpoint inhibitor nivolumab (Opdivo) vyráběný společností Bristol Myers Squibb (BMS) schválen v Evropské unii jako monoterapie pokročilého metastazujícího melanomu, k adjuvantní léčbě melanomu, po kompletní resekci nemalobuněčného karcinomu plic, renálního karcinomu, skvamózního karcinomu hlavy a krku, uroteliálního karcinomu a klasického Hodgkinova lymfomu. V kombinaci s dalším inhibitorem tzv. kontrolního bodu imunitní reakce ipilimumabem (Yervoy) je nivolumab navíc schválen k léčbě melanomu a renálního karcinomu.
V České republice je nivolumab hrazen v monoterapii melanomu (1. linie), nemalobuněčného karcinomu plic (2. a vyšší linie), renálního karcinomu (2. a 3. linie) a u klasického Hodgkinova lymfomu (po transplantaci vlastních krvetvorných kmenových buněk a po léčbě brentuximab vedotinem).
Mechanismus účinku
Léčivá látka selektivně blokuje
interakci mezi PD 1 a PD L1/PD L2 a tímto
způsobem restartuje imunitní reakci proti nádorovým buňkám.
Receptor programované buněčné smrti 1 ze skupiny antigenů
CD28 je součástí rozsáhlé a komplexní imunitní synapse
regulující imunitní odpověď [1,2]. Jeho úkolem je postupně
utlumit imunitní reakci vůči různým antigenům (bakteriím,
virům nebo nádorovým buňkám). Je exprimován na regulačních,
aktivovaných a paměťových T lymfocytech. Tento úkol je
navozen vazbou s PD L1 a PD L2, jež jsou
exprimovány na aktivovaných T lymfocytech, B lymfocytech,
na antigen prezentujících buňkách (dendritické buňky)
a na buňkách tvořících stroma a mikroprostředí
nádoru (tumorózní makrofágy, fibroblasty). Nádorové buňky,
které jsou vybaveny PD L1, mohou prostřednictvím tohoto
receptoru vypnout imunitní reakci cytotoxických T lymfocytů vůči
nim a znemožnit tak imunitní dohled nad nádorovými buňkami
[3]. Nivolumab přeruší tuto negativní zpětnou vazbu a zabrání
utlumení imunitní reakce (obr. 1).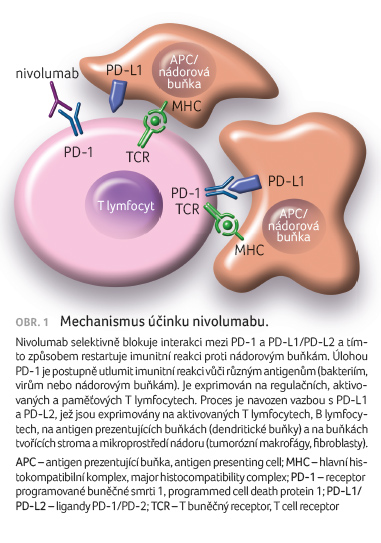
Farmakokinetické vlastnosti a údaje
o interakcích
Nivolumab je lidská monoklonální protilátka a její metabolismus nesouvisí s enzymy, které běžně metabolizují léčiva včetně nejčastěji používaného cytochromu P450. Nevyvolává inhibici ani indukci těchto enzymů. Také se nepředpokládá, že by jiné léčivé přípravky ovlivňovaly farmakokinetiku nivolumabu. Ani vliv systémových imunosupresivních kortikosteroidů nebyl v průběhu léčby prokázán. Jedinou možnou interakcí je ovlivnění farmakodynamiky nivolumabu u pacientů, jimž byly podány systémové kortikosteroidy a jiná imunosupresiva před zahájením léčby. Je zde potenciální možnost interference s mechanismem působení nivolumabu, nikoliv však s jeho farmakokinetikou. Nebyly provedeny žádné studie, které by sledovaly inkompatibilitu s jinými látkami. Nivolumab by měl být podáván v samostatné infuzi trvající 30 minut (240 mg každé dva týdny) nebo 60 minut (480 mg každé čtyři týdny). Farmakokinetika nivolumabu je lineární v rozpětí dávek 0,1‒10 mg/kg. Populační farmakokinetická analýza podávání nivolumabu 3 mg/kg 1× za dva týdny stanovila geometrický průměr clearance 9,5 ml/h, terminální biologický poločas 26,7 dne a míru průměrné expozice v ustáleném stavu 75,3 μg/ml. Clearance nivolumabu se zvyšuje s rostoucí tělesnou hmotností. V rozmezí tělesné hmotnosti 34‒162 kg je při standardní dávce nivolumabu hodnota minimální koncentrace stabilní.
Klinické zkušenosti
Melanom
Ve studii CheckMate 037 (NCT01721746, CA209 037) [4] byla hodnocena účinnost a bezpečnost nivolumabu proti chemoterapii dle výběru zkoušejícího u pacientů předléčených ipilimumabem nebo BRAF inhibitory (u pacientů s BRAF V600 mutovaným melanomem). Pacienti byli randomizováni v poměru 2 : 1 do ramene s dávkou nivolumabu 3 mg/kg podávanou každé dva týdny nebo do ramene s chemoterapií (dakarbazin 1 000 mg/m2 každé tři týdny, nebo paklitaxel 175 mg/m2 v kombinaci s karboplatinou AUC 6 [plocha pod křivkou plazmatické koncentrace] každé tři týdny). Léčba byla podávána do progrese choroby nebo do projevů intolerance terapie. Primárním cílovým ukazatelem studie byly počet objektivních odpovědí (objective response rate, ORR) a celkové přežití (overall survival, OS). Analýza ORR byla provedena po zařazení 120 pacientů do ramene s nivolumabem a s délkou sledování nejméně 24 týdnů. Potvrzené ORR byly zaznamenány u 38 (31,7 %, 95% CI [interval spolehlivosti] 23,5‒40,8) z prvních 120 pacientů ve skupině s nivolumabem a u pěti (10,6 %, 95% CI 3,5‒23,1) ze 47 pacientů ve skupině s chemoterapií. Závažné nežádoucí účinky 3. a 4. stupně, které souvisely s léčbou, byly zaznamenány u 12 (5 %) pacientů léčených nivolumabem a u devíti (9 %) pacientů ve skupině s chemoterapií. K žádnému úmrtí souvisejícímu s terapií nedošlo.
V randomizované dvojitě zaslepené studii fáze III CheckMate 066 (NCT01721772, CA209066) [5] byl porovnán účinek nivolumabu proti dakarbazinu u nepředléčených pacientů. Bylo randomizováno 418 pacientů v poměru 1 : 1 s neresekovatelným a metastatickým BRAF nemutovaným melanomem. Nivolumab byl podáván v dávce 3 mg/kg každé dva týdny a dakarbazin s placebem každé tři týdny, nebo dakarbazin v dávce 1 000 mg/m2 každé tři týdny a nivolumab s placebem každé dva týdny. Primárním cílovým ukazatelem studie byla doba OS. Jednoletého OS dosáhlo 72,9 % pacientů léčených nivolumabem a 42,1 % pacientů s léčbou dakarbazinem (HR [poměr rizik] 0,42; 95% CI 0,34‒0,56; p < 0,001). Medián přežití do progrese (progression free survival, PFS) představoval 5,1 měsíce ve skupině s nivolumabem proti 2,2 měsíce ve skupině s dakarbazinem (HR 0,43; 95% CI 0,34‒0,56; p < 0,001). Počet ORR dosáhl u nivolumabu 40 % a u dakarbazinu 13,9 % (p < 0,001). Léčba nivolumabem byla u všech definovaných podskupin pacientů, včetně hodnocení míry exprese PD L1, prospěšná oproti dakarbazinu. Nebyl prokázán rozdíl ve výsledcích u pacientů s vysokou a nízkou expresí receptorů PD L1. Závažné nežádoucí účinky stupně 3 a 4 související s léčbou byly prokázány u 11,7 % pacientů léčených nivolumabem a u 17,6 % pacientů léčených dakarbazinem.
Klinická studie fáze II CheckMate 069 (CA 209069) [6,7] přímo porovnala kombinaci nivolumab a ipilimumab se samotným podáváním ipilimumabu u pacientů s nepředléčeným neresekovatelným a metastazujícím melanomem BRAF V600 divokého typu. Bylo randomizováno 142 pacientů v poměru 2 : 1. Nemocní v rameni s kombinovanou terapií byli léčeni nivolumabem v dávce 1 mg/kg a ipilimumabem v dávce 3 mg/kg i.v. každé tři týdny po dobu podávání čtyř dávek, pak nivolumabem v dávce 3 mg/kg jednou za dva týdny až do progrese onemocnění nebo do nepřijatelné toxicity terapie. Pacienti v rameni s ipilimumabem obdrželi ipilimumab v dávce 3 mg/kg a placebo označené jako nivolumab intravenózně každé tři týdny po dobu podávání čtyř dávek, následně s placebem 1× za dva týdny. Při progresi onemocnění u pacientů v rameni s ipilimumabem byla nemocnému nabídnuta léčba nivolumabem v dávce 3 mg/kg každé dva týdny. U 109 pacientů s kombinovanou léčbou dosáhl počet ORR 60 % ve srovnání s 11 % u samotného ipilimumabu. Počet ORR se zvýšil o 49 % (95% CI 31‒61; p < 0,001). U 17 % pacientů léčených kombinací byla zaznamenána kompletní odpověď proti žádné odpovědi u nemocných léčených ipilimumabem. U skupiny pacientů s BRAF V600 mutací dosahoval počet ORR 52 % u kombinované léčby proti 10 % u samotného ipilimumabu. Důležité je, že obě skupiny pacientů, jak PD L1 pozitivní, tak PD L1 negativní, vykazovaly podobnou míru ORR (58 % vs. 55 %).
Výsledky studie fáze III CheckMate 067 (NCT01844505, CA209 067) [8] potvrzují závěry předchozí studie fáze II CheckMate 069. Celkem 945 pacientů s neléčeným neresekovatelným nebo metastazujícím melanomem bylo randomizováno do tří skupin: monoterapie nivolumabem (316 pacientů), monoterapie ipilimumabem (315 pacientů), nebo kombinace nivolumabu s ipilimumabem s pokračováním léčby nivolumabem (314 pacientů). V monoterapii byl nivolumab podáván v dávce 3 mg/kg jednou za dva týdny a ipilimumab v dávce 3 mg/kg každé tři týdny. V kombinované léčbě byly podávány nivolumab v dávce 1 mg/kg a ipilimumab v dávce 3 mg/kg každé tři týdny po dobu čtyř dávek a následně pokračovalo podávání nivolumabu v dávce 3 mg/kg každé dva týdny. Pacienti byli stratifikováni podle metastatického stadia, stavu PD L1 a stavu BRAF mutace. Přibližně jedna třetina pacientů vykazovala pozitivní BRAF V600 mutaci. Při více než devítiměsíčním sledování dosáhl medián PFS 11,5 měsíce při podávání kombinace, 6,9 měsíce při monoterapii nivolumabem a 2,9 měsíce u monoterapie ipilimumabem. Počet celkových ORR dosáhl 50 % u kombinace ve srovnání se 40 % u nivolumabu a se 14 % u ipilimumabu, resp. v rameni s kombinovanou léčbou bylo 8,9 % kompletních odpovědí, 8,5 % odpovědí pak v rameni s nivolumabem a 1,9 % odpovědí na léčbu v rameni s ipilimumabem. Významná toxicita (všechny stupně) byla vyjádřena nejvíce ve skupině pacientů s kombinovanou léčbou proti monoterapii nivolumabem a ipilimumabem, nejčastější nežádoucí účinky představovaly průjem (44,1 %, 19,2 %, 33,1 %), vyrážka (40,3 %, 25,9 %, 32,8 %), únava (35 %, 34,2 %, 28,0 %), svědění (33,2 %, 18,8 %, 35,4 %) a nevolnost (25,9 %, 13,1 %, 16,1 %). Toxicita stupně 3 a 4 byla zaznamenána u kombinované léčby v 55 %, u nivolumabu v 16,3 % a u ipilimumabu v 27,3 %. Nejčastěji uváděnými nežádoucími účinky byly průjem (9,3 %, 2,2 %, 6,1 %), kolitida (7,7 %, 0,6 %, 8,7 %), zvýšená hodnota lipázy (8,6 %, 3,5 %, 3,9 %), zvýšená hodnota alaninaminotransferázy (8,3 %, 1,3 % a 1,6 %) a zvýšená hodnota aspartátaminotransferázy (6,1 %, 1,0 %, 1,6 %). Vysoká účinnost kombinované léčby je vykoupena vysokou toxicitou. Definitivní výsledky OS nebyly dosud zveřejněny.
Nivolumab prokázal celkové pětileté přežití ve výši 34 % u těžce předléčených pacientů s metastatickým melanomem, kteří nebyli léčeni ipilimumabem. Výsledky studie fáze I byly předneseny na výročním zasedání American Association for Cancer Research (AACR) v dubnu 2016 [9]. Závěry studie CA209 003 představují první zprávu o dlouhodobých výsledcích klinické studie zkoumající účinek inhibitoru PD 1 a naznačují dlouhodobé přežití nemocných léčených nivolumabem v monoterapii. Do studie bylo zařazeno 107 pacientů, kteří byli léčeni nivolumabem podávaným každé dva týdny. Nemocní byli rozděleni do pěti skupin s postupným navyšováním dávky od 0,1 mg/kg do 10 mg/kg a pokračovali v léčbě do doby ≤ 96 týdnů. Primárním cílovým ukazatelem studie byly bezpečnost a snášenlivost, druhotným ukazatelem účinnost a změnu oproti původnímu protokolu znamenalo i posouzení dlouhodobého OS. Střední věk pacientů činil 61 let, performance status (PS) 0‒1 mělo 97 % osob, 62 % nemocných užívalo ≥ 2 předchozí terapie (včetně 46 %, kteří byli léčeni interleukinem 2). Při zahájení léčby mělo celkově 78 % pacientů viscerální metastázy a 36 % zvýšenou hodnotu laktátdehydrogenázy. Po minimální době sledování 45 měsíců všech skupin pacientů dosáhl medián OS 17,3 měsíce (95% CI 12,5‒37,8) a třicetiměsíčního PFS docílilo 18,6 % nemocných. U pacientů (v počtu 17) léčených dávkou nivolumabu 3 mg/kg (současná schválená dávka pro terapii nivolumabem) byl medián OS 20,3 měsíce (95% CI 7,2‒NR [nebylo dosaženo]). Pětiletého celkového přežití OS dosáhlo 35,3 % pacientů a třicetiměsíční PFS bylo zaznamenáno u 25,7 % osob.
Adjuvantní léčba melanomu
Ve studii CheckMate 238 (NCT02388906, CA209 238) [10] byla hodnocena účinnost a bezpečnost nivolumabu proti vysokodávkovanému ipilimumabu (10 mg/kg) u pacientů s kompletně resekovaným melanomem. Pacienti byli randomizováni v poměru 1 : 1 do ramene s nivolumabem v dávce 3 mg/kg podávané každé dva týdny nebo do ramene s ipilimumabem v dávce 10 mg/kg podávané každé tři týdny pro první čtyři dávky a dále každých 12 týdnů počínaje týdnem 24. Léčba byla podávána do progrese nemoci nebo do intolerance terapie maximálně 12 měsíců. Primárním cílovým ukazatelem studie bylo přežití bez recidivy (relaps free survival, RFS). Analýza RFS byla provedena po následném sledování přibližně 24 měsíců. Přežití bez recidivy ve 12 měsících bylo zaznamenáno u 70,4 % (95% CI 65,9‒74,4) pacientů ve skupině s nivolumabem a u 60,0 % (95% CI 55,2‒64,5) pacientů ve skupině s ipilimumabem. V 18 měsících u 65,8 % (95% CI 61,2‒70,0) oproti 53,0 % (95% CI 48,1‒57,6) a ve 24 měsících u 62,6 % (95% CI 57,9‒67,0) oproti 50,2 % (95% CI 45,3‒54,8) pacientů. Přínos v RFS byl prokázán konzistentně napříč podskupinami, včetně nádorové PD L1 exprese, BRAF statusu a stadia onemocnění.
Renální karcinom
Bezpečnost a účinnost nivolumabu byla prokázána u renálního karcinomu v otevřené randomizované studii fáze III CheckMate 025 (NCT01668784) [11]. Do studie bylo zařazeno 821 pacientů s pokročilým světlobuněčným renálním karcinomem, jejichž onemocnění se zhoršilo v průběhu léčby nebo po jejím přerušení po jedné až dvou předchozích liniích antiangiogenní terapie. Pacienti byli randomizováni v poměru 1 : 1. Nivolumab podávaný v dávce 3 mg/kg tělesné hmotnosti intravenózně každé dva týdny byl porovnán s everolimem podávaným v tabletách 10 mg perorálně jednou denně. Primárním cílovým ukazatelem studie byla délka OS a sekundárními cílovými ukazateli byly ORR a bezpečnost. Medián OS při léčbě nivolumabem dosáhl 25,0 měsíce (95% CI, 21,8‒NR) a při léčbě everolimem 19,6 měsíce (95% CI, 17,6‒23,1), HR 0,73 (98,5% CI 0,57‒0,93; p = 0,002). Počet objektivních odpovědí byl vyšší u pacientů léčených nivolumabem (25 %) oproti everolimu (5 %; p < 0,001), medián doby trvání odpovědi byl 11,99 měsíce v obou ramenech. Medián PFS dosáhl u nivolumabu 4,6 měsíce a u everolimu 4,4 měsíce a rozdíl byl statisticky nevýznamný (HR 0,88; 95% CI 0,75‒1,03; p = 0,11). Nežádoucí účinky 3. nebo 4. stupně se vyskytly u 19 % pacientů užívajících nivolumab a u 37 % pacientů užívajících everolimus. Nejčastějším nežádoucím efektem u nivolumabu byla únava (2 % nemocných) a u everolimu anémie (8 % nemocných).
Bezpečnost a účinnost kombinace nivolumabu a ipilimumabu byla prokázána u renálního karcinomu v otevřené randomizované studii fáze III CheckMate 214 (NCT02231749) [12]. Do studie bylo zařazeno 1 096 pacientů s dosud neléčeným pokročilým světlobuněčným renálním karcinomem. Pacienti byli randomizováni v poměru 1 : 1. Nivolumab podávaný v dávce 3 mg/kg v kombinaci s ipilimumabem v dávce 1 mg/kg intravenózně každé tři týdny u prvních čtyř dávek a následně nivolumab v dávce 3 mg/kg v monoterapii každé dva týdny byl porovnán se sunitinibem podávaným v tabletách 50 mg perorálně denně čtyři týdny s následnou dvoutýdenní přestávkou. Léčba trvala, dokud byl pozorován klinický přínos nebo dokud byla terapie tolerována. Primárním cílovým ukazatelem studie byly délka OS, ORR a PFS určené nezávislou centrální komisí u pacientů se středním/vysokým rizikem. Mediánu OS u kombinace nivolumabu a ipilimumabu nebylo dosaženo (95% CI 28,2‒NE [neodhadnutelné]), u sunitinibu činil 25,9 měsíce (95% CI 22,1‒NE), HR 0,63 (98,5% CI 0,44‒0,89; p = 0,0001). Počet potvrzených objektivních odpovědí byl signifikantně vyšší u pacientů léčených kombinací nivolumab a ipilimumab (41,6 %) oproti sunitinibu (26,5 %; p < 0,0001). Medián PFS dosáhl u kombinace nivolumabu a ipilimumabu 11,6 měsíce (95% CI 8,7‒15,5), u sunitinibu 8,4 měsíce a rozdíl nebyl statisticky nevýznamný (HR 0,82; 95% CI 0,64‒1,05; p = 0,0331).
Nemalobuněčný karcinom plic
Možnosti léčby skvamózního nemalobuněčného karcinomu plic (non small cell lung cancer, NSCLC) jsou omezené. V randomizované otevřené studii fáze III CheckMate 017 (NCT01642004) [13] byl porovnán účinek nivolumabu s docetaxelem ve druhé linii léčby po selhání první linie kombinační chemoterapie na bázi platinového derivátu. Pacienti (v počtu 272) byli randomizováni v poměru 1 : 1. V rameni s nivolumabem v dávce 3 mg/kg 1× za dva týdny bylo zařazeno 135 pacientů a v rameni s docetaxelem v dávce 75 mg/m2 1× za tři týdny 137 pacientů. Léčba probíhala do progrese onemocnění nebo do intolerance terapie. Studie splnila primární cílový ukazatel. V parametru OS dosáhl medián u nivolumabu 9,2 měsíce oproti docetaxelu s délkou 6,0 měsíce (HR 0,59; 95% CI 0,44‒0,79; p = 0,00025). Riziko úmrtí bylo u pacientů s nivolumabem sníženo o 41 %. Jednoletého přežití dosáhlo s nivolumabem 42 % pacientů oproti 24 % s docetaxelem. Také v ukazateli PFS byl nivolumab lepší. Medián PFS u nivolumabu dosáhl 3,5 měsíce oproti docetaxelu s mediánem 2,8 měsíce (HR 0,62; 95% CI 0,47‒0,81; p = 0,0004) a jednoročního PFS dosáhlo 20,8 % pacientů s nivolumabem oproti 6,4 % pacientů s docetaxelem. Celkový ORR byl 20 % (27/135) u léčených s nivolumabem oproti 9 % (12/137) s docetaxelem (p = 0,0083). Stanovená hodnota exprese PD L1 neprokázala prediktivní ani prognostický význam. Studie zahrnovala rovněž hodnocení kvality života a bezpečnost podání. Stupeň 3 a 4 u nežádoucích účinků byl zaznamenán v 7 % (9/131) u nivolumabu a v 55 % (71/129) u docetaxelu. Nebyla zaznamenána žádná úmrtí, která by byla spojena s léčbou nivolumabem, a byla zaznamenána tři úmrtí při terapii docetaxelem. Bezpečnostní profil nivolumabu se ukázal být lepším než v případě docetaxelu.
U pacientů s neskvamózním NSCLC prokázal nivolumab podobné výsledky v randomizované otevřené studii fáze III CheckMate 57 (NCT01673867) [14]. Do studie byli v poměru 1 : 1 randomizováni pacienti, kteří byli v první linii předléčeni chemoterapií s platinovými deriváty nebo tyrozinkinázovým inhibitorem. Do studie bylo zařazeno celkem 292 pacientů léčených nivolumabem (3 mg/kg 1× za dva týdny i.v.) a 290 pacientů s docetaxelem (75 mg/m2 1× za tři týdny i.v.). Celkové přežití bylo výrazně prodlouženo u pacientů s nivolumabem ‒ medián 12,2 měsíce oproti 9,4 měsíce při léčbě docetaxelem (HR 0,73; 96% CI 0,59‒0,89; p = 0,00155). Ročního OS dosáhlo 50,5 % pacientů léčených nivolumabem a 39,0 % pacientů léčených docetaxelem. Také bylo dosaženo zlepšení ORR (19,2 % vs. 12,4 %; p = 0,0235). Riziko úmrtí a progrese se snížilo u pacientů s nivolumabem o 27 %. V této studii byla exprese PD L1 spojena s lepšími výsledky léčby nivolumabem a účinnost terapie byla prokázána napříč všemi hodnotami receptorů PD L1 (1 %, 5 % a 10 %). Stupeň 3‒5 nežádoucích účinků souvisejících s léčbou nivolumabem se vyskytoval u 10,5 % (30/287) pacientů a u 53,7 % (144/268) pacientů léčených docetaxelem. V souvislosti s léčbou nivolumabem se neprokázalo žádné úmrtí proti jednomu úmrtí u pacientů s docetaxelem.
Prediktivní a prognostické
faktory
Prediktivní faktor pro léčbu anti PD 1 protilátkou se stále hledá. Jako logický kandidát se jeví koncentrace jeho ligandu ‒ PD L1. Jeho přítomnost je možné prokázat na všech buňkách, které tvoří nádor, včetně jeho stromatu. Také kvantifikace exprese PD L1 v nádorovém vzorku vyžaduje určení vhodné metody. Ve studiích s léčbou inhibitory anti PD 1 či anti PD L1 byly u NSCLC nejčastěji používány tři metody: Ventana SP263, Dako 22C3 a Dako 28 8. Bylo vyhodnoceno 500 vzorků NSCLC [13]. Klinické a demografické charakteristiky pacientů, jejichž vzorky nádorů se použily k porovnání jednotlivých diagnostických metod, byly relativně rovnoměrné a zahrnovaly pacienty s věkovým rozložením mladší než 60 let vs. 60 a více let, 76 % osob mužského pohlaví, 80 % osob se stadiem onemocnění I‒II, 54 % osob s histologií adenokarcinomů a 43 % osob s histologií dlaždicobuněčných karcinomů. Korelace membránového barvení mezi metodami Ventana SP263 a Dako 22C3 byla 91 %. Korelace mezi Ventana SP263 a Dako 28 8 činila 93,5 % a mezi Dako 28 8 a Dako 22C3 95 %. Nalezení optimálního hodnocení pozitivity PD L1 je nutné k ověření váhy predikce tohoto receptoru. Bohužel výsledky ze studií u melanomu neprokázaly souvislost koncentrace PD L1 s účinností léčby nivolumabem. Stejné výstupy byly pozorovány ve studii CheckMate 025 s léčbou renálního karcinomu, kde byl výsledek terapie nivolumabem nezávislý na koncentraci receptorů PD L1. V léčbě NSCLC jsou výsledky rozdílné. U skvamózního NSCLC ve studii CheckMate 017 se hodnota PD L1 neukázala jako prediktor účinnosti léčby na rozdíl od studie CheckMate 057 s neskvamózním NSCLC, kde byl prediktivní význam exprese PD L1 prokázán. Tyto rozdílné výsledky vyvolávají pochybnosti o možnosti využití koncentrace PD L1 receptorů jako prediktoru účinnosti nivolumabu napříč nádory. Možným vysvětlením tohoto kolísání hodnoty PD L1 je to, že jeho exprese je výsledkem komplexní reakce zahrnující zánětlivou a autoimunitní složku ve stromatu nádoru. Hlavním mediátorem je interferon gama (IFNγ). Podílí se na regulaci téměř všech fází imunitních a zánětlivých reakcí. Aktivuje růst a diferenciaci T lymfocytů, B lymfocytů, nádorových makrofágů, NK buněk a buněk, které tvoří stroma nádoru ‒ endoteliálních buněk a fibroblastů. Zesiluje expresi hlavního histokompatibilního komplexu (major histocompatibility complex, MHC) II na antigen prezentujících buňkách a zvyšuje protinádorový účinek lymfocytů. Podařilo se identifikovat skupinu genů (tzv. gene signature) spojenou se zánětlivou a imunitní reakcí řízenou IFNγ, jejichž přítomnost je spojena s výsledky léčby anti PD 1 u pacientů s melanomem [16]. Produkty této skupiny genů zajišťují signální dráhy IFNγ, prezentaci antigenů a tvorbu T lymfocytárních markerů. Výsledky potvrdily předpoklad, že účinnost blokády PD 1 se objevuje u pacientů s preexistující adaptivní imunitní odpovědí řízenou IFNγ. Takzvaná IFNγ gene signature je možným budoucím biomarkerem pro selekci pacientů k léčbě anti PD 1 protilátkou, pokud se podaří tyto výsledky potvrdit v prospektivních studiích.
Bezpečnost a snášenlivost
Nejběžnějšími nežádoucími účinky terapie nivolumabem jsou únava, slabost nebo nedostatek energie (astenické stavy), kašel, nevolnost, vyrážka, dýchací potíže (dušnost), průjem, zácpa, snížená chuť k jídlu, bolesti zad a kloubů (artralgie). Nivolumab má rovněž potenciál způsobit závažné nežádoucí účinky, které vyplývají z jeho schopnosti ovlivnit imunitní systém (známé jako tzv. imunitně zprostředkované nežádoucí účinky). Tyto těžké imunitně zprostředkované nežádoucí účinky mohou poškodit důležité zdravé orgány: plíce, tlusté střevo, játra, ledviny, žlázy produkující hormony a mozek [17].
Pneumonitida nebo intersticiální onemocnění plic je závažným nežádoucím efektem a byly zaznamenány i fatální případy. Při léčbě nivolumabem by měli být pacienti v případě dechových obtíží monitorováni rentgenologickým vyšetřením, které může odhalit příznaky jako ohniskové opacity mléčného skla a ložiskové filtráty. Vždy musejí být vyloučeny infekční příčiny. Klinickými projevy jsou dušnost a hypoxie. Při zaznamenání symptomatické pneumonitidy 2. stupně musí být léčba nivolumabem přerušena a měla by být zahájena terapie kortikosteroidy v dávce odpovídající 1 mg/kg metylprednisolonu denně. Po zlepšení lze znovu podat nivolumab po postupném utlumení užívání kortikosteroidů, které musí probíhat alespoň po dobu čtyř týdnů. Jestliže dojde ke zhoršení nebo se nedostaví zlepšení navzdory užití kortikosteroidů, lze dávku zvýšit na odpovídající 2‒4 mg/kg metylprednisolonu denně a musí být trvale ukončeno podávání nivolumabu. U pneumonitidy 3. nebo 4. stupně musí být ukončeno podávání nivolumabu trvale a má být zahájena léčba kortikosteroidy v dávce odpovídající 2‒4 mg/kg metylprednisolonu denně.
Imunitně podmíněná kolitida při léčbě nivolumabem se projevuje závažnými průjmy a dalšími symptomy, jako jsou bolest břicha, hlen a krev ve stolici. Pokud se vyskytnou takové symptomy při léčbě nivolumabem, musejí být pacienti monitorováni a musí být vyloučena infekční příčina. U průjmu nebo kolitidy 2. stupně má být podávání nivolumabu ukončeno. Pokud po ukončení nedojde k odeznění symptomů, je nutné podat kortikosteroidy v dávce odpovídající 0,5‒1 mg/kg metylprednisolonu denně. Po zlepšení lze znovu podat nivolumab po postupném utlumení užívání kortikosteroidů. Jestliže dojde ke zhoršení nebo se nedostaví zlepšení, je nutno podávání nivolumabu trvale ukončit. U průjmu nebo kolitidy 3. stupně má být léčba nivolumabem ukončena a mají být podány kortikosteroidy v dávce odpovídající 1‒2 mg/kg metylprednisolonu denně. Po zlepšení lze znovu podat nivolumab po postupném utlumení léčby kortikosteroidy. Jestliže dojde ke zhoršení nebo se nedostaví zlepšení přes použití kortikosteroidů, podávání nivolumabu musí být trvale ukončeno. U průjmu nebo kolitidy 4. stupně musí být trvale ukončeno podávání nivolumabu a má být zahájena léčba kortikosteroidy v dávce odpovídající 1‒2 mg/kg metylprednisolonu denně.
Imunitně podmíněná hepatitida se projevuje příznaky hepatitidy, jako je zvýšení hodnoty transamináz a celkového bilirubinu, ale bez průkazu infekční etiologie a etiologie spojené se základním onemocněním. Pokud dojde ke zvýšení hodnoty transamináz nebo celkového bilirubinu 2. stupně, doporučuje se podávání nivolumabu přerušit. Nedojde li ke spontánnímu odeznění příznaků, musejí být podány kortikosteroidy v dávce odpovídající 0,5‒1 mg/kg metylprednisolonu denně. Po úpravě parametrů lze v podávání nivolumabu pokračovat. Pokud potíže přetrvávají nebo se zhorší, je nutno léčbu nivolumabem trvale ukončit. Při zvýšení hodnoty transamináz nebo celkového bilirubinu 3. a 4. stupně musí být podávání nivolumabu trvale ukončeno a má být zahájena léčba kortikosteroidy v dávce odpovídající 1‒2 mg/kg metylprednisolonu denně.
Imunitně podmíněná nefritida nebo renální dysfunkce se projevuje při léčbě nivolumabem asymptomatickým nárůstem koncentrace kreatininu v séru. Po vyloučení etiologie spojené se základním onemocněním a při zvýšení hodnoty sérového kreatininu 2. nebo 3. stupně má být podání nivolumabu přerušeno a zahájena léčba kortikosteroidy v dávce odpovídající 0,5‒1 mg/kg metylprednisolonu denně. Po zlepšení lze znovu podat nivolumab po postupném utlumení podávání kortikosteroidů. Jestliže dojde ke zhoršení nebo se nedostaví zlepšení přes použití kortikosteroidů, má být dávka kortikosteroidů zvýšena na dávku odpovídající 1‒2 mg/kg metylprednisolonu denně a podávání nivolumabu musí být trvale ukončeno. Při zvýšení sérové koncentrace kreatininu 4. stupně musí být podávání nivolumabu trvale ukončeno a má být zahájena léčba kortikosteroidy v dávce odpovídající 1‒2 mg/kg metylprednisolonu denně.
Imunitně podmíněná endokrinopatie se projevuje nejčastěji jako hypotyreóza, hypofyzitida, funkční nedostatečnost nadledvin a diabetická ketoacidóza. Vzhledem k očekávání projevů této toxicity musejí být pacienti při léčbě nivolumabem monitorováni před léčbou a v jejím průběhu pomocí biochemických parametrů. Klinicky se projevují únavou, bolestmi hlavy, změnami duševního stavu, bolestmi břicha, neobvyklými střevními projevy a hypotenzí nebo nespecifickými symptomy, které se mohou podobat přítomnosti mozkových metastáz. Po vyloučení příznaků spojených s vlastním generalizovaným onemocněním je lze považovat za imunitně podmíněnou endokrinopatii. U symptomatické hypotyreózy má být podání nivolumabu přerušeno a dle potřeby má být zahájena substituční léčba hormony štítné žlázy. U symptomatické hypertyreózy je kromě ukončení podávání nivolumabu dle potřeby zahájena léčba methimazolem. Pokud existuje podezření na akutní zánět štítné žlázy, má být zváženo podání kortikosteroidů v dávce odpovídající 1‒2 mg/kg metylprednisolonu denně. Po zlepšení lze znovu podat nivolumab po postupném utlumení podávání kortikosteroidů, je li třeba. Monitorování funkce štítné žlázy má i nadále pokračovat, aby byla zajištěna odpovídající hormonální substituční léčba. Při symptomatické nedostatečnosti nadledvin po přerušení podávání nivolumabu je žádoucí dle potřeby zahájit léčbu kortikosteroidy a je nutné nadále monitorovat hormonální parametry a dle toho zajistit odpovídající substituční léčbu. V případě symptomatické hypofyzitidy se po přerušení podávání nivolumabu musí zahájit substituční hormonální léčba. Pokud existuje podezření na akutní zánět hypofýzy, je nutno zvážit podání kortikosteroidů v dávce odpovídající 1‒2 mg/kg metylprednisolonu denně. Pokud dojde ke zlepšení stavu, lze podat nivolumab po postupném utlumení užívání kortikosteroidů. Nadále je nutné monitorovat funkce hypofýzy a sledovat hormonální parametry k zajištění odpovídající hormonální substituční léčby. U symptomatického diabetu je po přerušení podávání nivolumabu dle nálezů nutné zahájit substituční léčbu inzulinem. Monitorování hodnoty glukózy zajistí správnou substituční léčbu inzulinem.
Imunitně zprostředkovaná kožní toxicita se projevuje různými typy vyrážky. Vyrážka je smíšený pojem, který zahrnuje vyrážku makulopapulární, erytematózní, svědivou, folikulární, makulární, papulární, pustulární, vezikulární, dermatitidu, akneiformní dermatitidu, alergickou dermatitidu a exfoliativní dermatitidu. Podávání nivolumabu má být přerušeno při vyrážce 3. stupně a ukončeno při stupni 4. Závažnou vyrážku léčíme vysokými dávkami kortikosteroidů odpovídajícími dávce 1‒2 mg/kg/den prednisonu. Opatrnosti při podání nivolumabu je třeba u pacientů, kteří prodělali závažné nebo život ohrožující kožní nežádoucí účinky při předchozí léčbě jinými imunostimulačními protinádorovými léky.
U méně než 1 % pacientů byly zaznamenány následující imunitně podmíněné nežádoucí účinky v souvislosti s léčbou nivolumabem (různé klinické studie, různé typy nádorů a odlišné dávkování léku): pankreatitida, uveitida, demyelinizace, autoimunitní neuropatie (včetně parézy n. facialis a n. abducens), syndrom Guillaina‒Barrého, hypopituitarismus a myastenický syndrom.
Pokud se vyskytnou závažné nežádoucí účinky léčby v průběhu aplikace infuze nivolumabu, je nutné terapii přerušit. V případě výskytu nežádoucích reakcí 4. stupně musí být léčba ukončena. V případě výskytu nežádoucích účinků nižších stupňů je terapie přerušena a po odeznění reakce se pokračuje za přísného monitorování v další léčbě.
Kontraindikace
Podání nivolumabu je kontraindikováno
v případě alergické reakce na účinnou látku nebo
na některou z pomocných látek uvedených v souhrnu
údajů o přípravku (SPC) [17].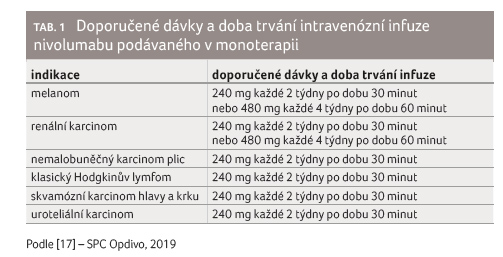
Popis léčivého přípravku
Nivolumab je dodáván jako koncentrát
pro infuze 1 mg v 10 ml [17]. Koncentrát je čirá až
opalizující bezbarvá až světle žlutá tekutina, která může
obsahovat několik světlých částic. Roztok má pH zhruba 6,0
a osmolalitu přibližně 340 mOsm/kg. Je dostupný
v baleních 40 mg, 100 mg a 240 mg. Doporučená
dávka přípravku je 240 mg/kg podávaných pouze intravenózně
po dobu 30 minut každé dva týdny, nebo 480 mg podávaných
pouze intravenózně po dobu 60 minut každé čtyři týdny.
Doporučené dávky nivolumabu v monoterapii v jednotlivých
indikacích a nivolumabu v kombinaci s ipilimumabem
shrnuje tabulka 1 a 2.
Léčba by měla pokračovat, dokud je pozorován klinický přínos
nebo dokud ji pacient toleruje. Zvyšovat nebo snižovat dávku se
nedoporučuje. Ukončení nebo přerušení léčby je závislé
na individuální bezpečnosti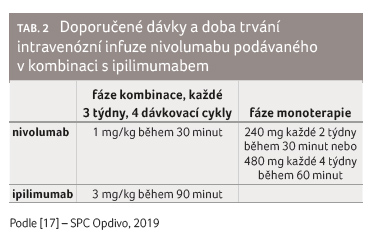 a snášenlivosti. Infuze se
musí podávat přes sterilní, nepyrogenní filtr s nízkou
schopností vázat proteiny a s póry o velikosti
0,2‒1,2 μm. Přípravek se nesmí podávat jako nitrožilní bolus
nebo bolusová injekce. Celková požadovaná dávka nivolumabu se
může podávat v infuzi přímo jako roztok s koncentrací
10 mg/ml, nebo může být naředěna až na 1 mg/ml
roztokem chloridu sodného 9 mg/ml (0,9 %) pro injekce nebo
roztokem glukózy 50 mg/ml (5 %) pro injekce. Přípravek musí
být uchováván v chladu (2–8 °C), nesmí zmrznout
a po naředění je stabilní 24 hodin při teplotě
2–8 °C a čtyři hodiny včetně času podání při
teplotě 20–25 °C. Po celou dobu musí být chráněn před
světlem.
a snášenlivosti. Infuze se
musí podávat přes sterilní, nepyrogenní filtr s nízkou
schopností vázat proteiny a s póry o velikosti
0,2‒1,2 μm. Přípravek se nesmí podávat jako nitrožilní bolus
nebo bolusová injekce. Celková požadovaná dávka nivolumabu se
může podávat v infuzi přímo jako roztok s koncentrací
10 mg/ml, nebo může být naředěna až na 1 mg/ml
roztokem chloridu sodného 9 mg/ml (0,9 %) pro injekce nebo
roztokem glukózy 50 mg/ml (5 %) pro injekce. Přípravek musí
být uchováván v chladu (2–8 °C), nesmí zmrznout
a po naředění je stabilní 24 hodin při teplotě
2–8 °C a čtyři hodiny včetně času podání při
teplotě 20–25 °C. Po celou dobu musí být chráněn před
světlem.
Závěr
Imunoterapie zasahuje do všech oblastí současné onkologické léčby. Jsme doslova zaplavováni informacemi o nových léčebných indikacích u molekul, jakou představuje nivolumab. Jeho široké uplatnění se odráží v tomto souhrnu, kde byly detailně popsány pouze ty diagnózy, v nichž nivolumab získal úhradu z veřejného zdravotního pojištění v České republice.
Seznam použité literatury
- [1] Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD‑1 and its ligands in tolerance and immunity. Annu Rev Immunol 2008; 26: 677‒704.
- [2] Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012; 12: 252‒264.
- [3] Swann JB, Smyth MJ. Immune surveillance of tumors. J Clin Invest 2007; 117: 1137‒1146.
- [4] Weber JS, DʼAngelo SP, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti‑CTLA‑4 treatment (CheckMate 037): a randomised, controlled, open‑label, phase 3 trial. Lancet Oncol 2015; 16: 375‒384.
- [5] Robert C, Long GV, Brady B. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015; 372: 320‒330.
- [6] Postow MA, Chesney J, Pavlick AC, et al. Initial report of overall survival rates from a randomized phase II trial evaluating the combination of nivolumab (NIVO) and ipilimumab (IPI) in patients with advanced melanoma (MEL). Presented at: AACR 2016, New Orleans; April 16‒20, 201. Abstract CT002.
- [7] Postow MA, Chesney J, Pavlick AC, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med 2015; 372: 2006‒2017.
- [8] Larkin J, Chiarion‑Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015; 373: 23‒34.
- [9] Hodi SF, Kluger HM, Sznol M, et al. Durable, Long‑term Survival in Previously Treated Patients With Advanced Melanoma Who Received Nivolumab Monotherapy in a Phase I Trial. Presented at the 2016 AACR Annual Meeting; April 16‒20, New Orleans, Louisiana. Abstract CT001.
- [10] Weber J, Mandala M, Del Vecchio M, et al. Adjuvant nivolumab versus ipilimumab in resected stage III or IV melanoma. N Engl J Med 2017; 377: 1824‒1835.
- [11] Motzer RJ, Escudier B, McDermott DF, et al. Nivolumab versus Everolimus in Advanced Renal‑Cell Carcinoma. N Engl J Med 2015; 373: 1803‒1813.
- [12] Motzer RJ, Tannir NM, McDermott DF, et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal‑cell carcinoma. N Engl J Med 2018; 378: 1277‒1290.
- [13] Spigel DR, Reckamp KL, Rizvi N, et al. A phase III study (CheckMate 017) of nivolumab (NIVO; anti‑programmed death‑1 [PD‑1]) vs docetaxel (DOC) in previously treated advanced or metastatic squamous (SQ) cell non‑small cell lung cancer (NSCLC). J Clin Oncol 2015; 33(Suppl; Abstr 8009).
- [14] Paz‑Ares L, Horn L, Borghaei H. Phase III, randomized trial (CheckMate 057) of nivolumab (NIVO) versus docetaxel (DOC) in advanced non‑squamous cell (non‑SQ) non‑small cell lung cancer (NSCLC). J Clin Oncol 2015; 33(Suppl); Abstr LBA109.
- [15] Ratcliffe MJ, Sharpe A, Midha A, et al. A comparative study of PD‑L1 diagnostic assays and the classification of patients at PD‑L1 positive and PD‑L1 negative. Presented at the 2016 AACR Annual Meeting; April 16‒20; New Orleans, Louisiana. Poster LB‑094.
- [16] Ribas A, Robert C, Hodi FS, et al. Association of response to programmed death receptor 1 (PD‑1) blockade with pembrolizumab (MK‑3475) with an interferon‑inflammatory immune gene signature. J Clin Oncol 2015; 33(Suppl); Abstr 3001.
- [17] Opdivo® (nivolumab). Souhrn informací o přípravku, 2019. Dostupné na: https://www.ema.europa.eu/documents/product‑information/opdivo‑epar‑product‑information_cs.pdf