Nové možnosti terapie cystické fibrózy
Souhrn:
Doušová T. Nové možnosti terapie cystické fibrózy. Remedia 2020; 30: 489–491.
Cystická fibróza (CF) představuje nejčastější život limitující dědičné onemocnění, které postihuje přibližně 85 000 pacientů po celém světe. Příčinou onemocnění je mutace v genu CFTR (cystic fibrosis conductance regulator), který plní funkci iontového přenašeče na apikální membráně epitelových buněk. Léčba tohoto závažného onemocnění byla donedávna pouze symptomatická. Možnosti kauzální terapie mířící přímo na opravu molekulárního defektu CFTR proteinu se objevily v roce 2012, kdy byl registrován první lék ze třídy tzv. CFTR modulátorů opravujících funkci tohoto proteinu. V současné době máme k dispozici čtyři CFTR modulátory, všechny dohromady mohou účinkovat až u 90 % pacientů s CF. Určujícím parametrem příslušné léčby je genotyp pacienta.
Summary:
Dousova T. New possibilities of cystic fibrosis treatment. Remedia 2020; 30: 489–491.
Cystic fibrosis (CF) is the most common life‑shortening genetic disease. It affects approximately 85,000 people worldwide. The disease is caused by bi‑allelic mutations in the gene encoding the CFTR (cystic fibrosis conductance regulator) protein, which plays a major role in ion transport across the apical membrane of the epithelial cells. Until recently, treatment of this disease was solely symptomatic. Causal therapy targeting molecular defect of CFTR protein has been available since 2012, when first therapeutic agent was registered. These therapeutic agents (CFTR modulators) repair synthesis and function of the CFTR protein. Currently, there are four CFTR modulators available and all together they can work in up to 90% of all patients with CF. Determining parameter of the respective treatment is the genotype of the patient.
Key words: cystic fibrosis, causal therapy, CFTR modulators
Úvod
Cystická fibróza (CF) je nejčastějším život limitujícím monogenně podmíněným onemocněním, postihujícím přibližně 85 000 pacientů na celém světě [1]. Příčinou je mutace v genu kódujícím chloridový kanál na apikální membráně epiteliálních buněk (cystic fibrosis transmembrane conductance regulator, CFTR). Typický fenotyp představuje progredující plicní onemocnění, ostatní symptomy CF zahrnují pankreatickou insuficienci, zvýšenou hodnotu chloridů v potu a v případě mužského pohlaví neplodnost. Limitujícím faktorem přežití zůstává plicní postižení, na jehož vzniku se spolu s obstrukcí dýchacích cest vazkým hlenem podílejí i chronické zánětlivé změny a mikrobiální kolonizace, a to i přes současné možnosti symptomatické (standardní) terapie [2,3].
Dlouhodobě všechny možnosti terapie CF představovaly léčbu symptomatickou: fyzioterapie, mukolytika, antibiotika, léky s protizánětlivým účinkem a léky vedoucí ke zlepšení stavu výživy pacienta. Na rozdíl od léčby kauzální však neřeší molekulární defekt samotného onemocnění, na jehož úrovni by bylo možné principiálně zabránit sérii nepříznivých změn vedoucích k nevratnému poškození plicní tkáně. Od roku 1989, kdy byl identifikován CFTR gen [4], je výzkum v oblasti CF zaměřen právě na vývoj nových terapií směřujících k opravě mutovaného genu, resp. k záchraně ztracené funkce CFTR proteinu.
Mutace genu CFTR
Do dnešního dne bylo popsáno více než 2 000 variant genu CFTR, z nichž jen u malého počtu (celkem ~300) byla jednoznačně prokázána kauzalita s onemocněním CF [5]. Naprostou většinu představuje mutace F508del přítomná u 90 % pacientů alespoň na jedné alele. Ze zbývajících mutací se pouze 20 vyskytuje ve frekvenci vyšší než 0,1 %, s určitou mírou variability vázanou na specifické populace (keltská mutace, slovanská mutace) [6]. Ke snazšímu pochopení patogenetického mechanismu vzniku defektního proteinu byly mutace CFTR rozděleny do sedmi funkčních tříd [3]. Mutace I. třídy (tzv. stop codon mutace) vedou k tvorbě zkráceného a nefunkčního proteinu. Mutace II. třídy dávají vzniknout strukturálně abnormálnímu proteinu, který nevycestuje intracelulárně k buněčné membráně (tzv. trafficking mutace). Při mutacích III. a IV. třídy protein vycestuje k povrchu buňky, avšak jeho funkce je alterována. V prvním případě ve smyslu nedostatečného otevírání kanálu (tzv. gating mutace), v případě druhém nedostatečným vedením iontů kanálem.
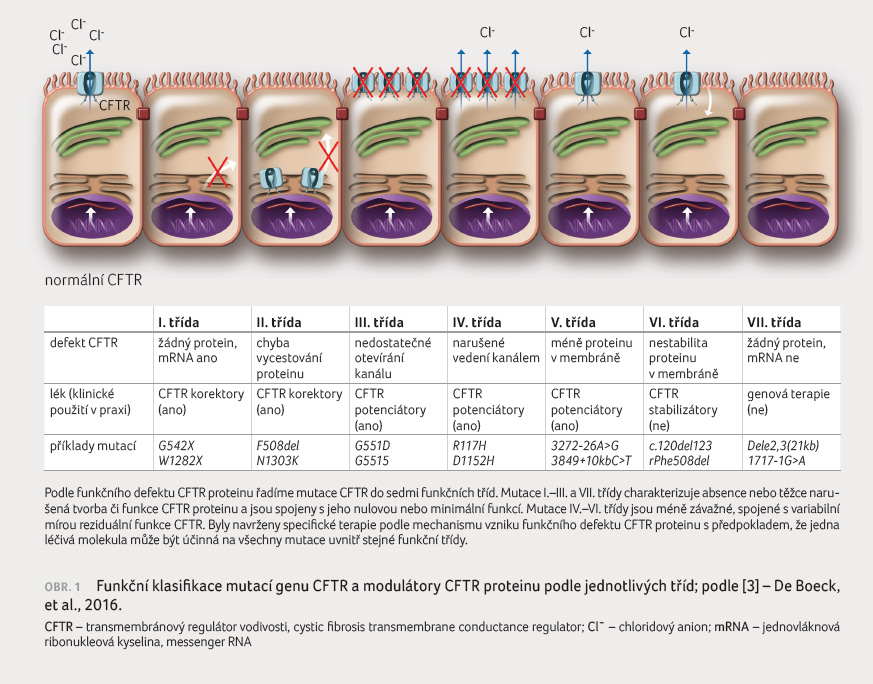
V. třída mutací (tzv. splicing mutace) vede k nedostatečné tvorbě CFTR proteinu a tím k jeho sníž
enému množství na povrchu buňky. Mutace VI. třídy vedou ke zkrácení životnosti proteinu kvůli jeho strukturální nestabilitě a konečně mutace VII. třídy, též klasifikované jako farmakologicky nezachranitelné, zahrnují velké deleční mutace, jejichž vlivem nedojde k tvorbě ani minimálního množství jednovláknové ribonukleové kyseliny (messenger RNA, mRNA)v buněčném jádře.
Uvedená klasifikace dnes současně napomáhá předurčit různý účinek nové terapie na různé třídy mutací (obr. 1) [3].
Modulátory CFTR proteinu – kauzální terapie cystické fibrózy
V roce 2012 byly registrovány první možnosti kauzální terapie CF, které v klinických studiích prokázaly zlepšení funkce plic, snížení hodnoty chloridů v potu, zlepšení stavu výživy a zlepšení jiných relevantních klinických parametrů v porovnání s placebem [7‒9].
Tyto léčebné molekuly označujeme jako tzv. modulátory CFTR proteinu a rozdělujeme je na potenciátory a korektory. Zatímco potenciátory zvyšují omezené otevírání CFTR kanálu a účinkují tak jen v situacích, kdy je protein přítomen na membránách buněk, korektory působí v kaskádě syntézy CFTR o krok dříve a upravují chybu intracelulárního vycestování špatně nařasené bílkoviny směrem k buněčné membráně. Modulátory CFTR proteinu mohou zmírnit ireverzibilní změny, nebo jim dokonce předejít, pokud je léčba zahájena před nástupem prvních symptomů onemocnění. Genotyp pacienta je určujícím parametrem příslušné léčby, která je podávána v dávce odvíjející se od věku nemocného.
Ivakaftor (KalydecoTM, Vertex Pharmaceuticals) byl vůbec prvním CFTR modulátorem (potenciátorem) schváleným v roce 2012 ke klinickému využití u pacientů nesoucích nejčastější mutaci III. třídy G551D [7]. Tento potenciátor byl později schválen pro dalších osm mutací stejné funkční třídy [10] a pro větší počet mutací s reziduální funkcí (např. D1152H, 3849+10kbC>T, 2789+5G>A z celkových 38), byť v jejich případě se prokázal vyšší účinek v kombinaci s korektorem [11]. V roce 2015 byla registrována první kombinace CFTR korektoru lumakaftoru s ivakaftorem (OrkambiTM, Vertex Pharmaceuticals), která vedla ke zlepšení hodnot plicních funkcí a ke snížení počtu akutních exacerbací u pacientů s mutací F508del v homozygotním stavu [8]. Obdobného efektu bylo dosaženo kombinací korektoru tezakaftoru s ivakaftorem (SymkeviTM, Vertex Pharmaceuticals), která byla schválena roku 2018 [9]. Přípravek Symkevi byl schválen pro léčbu dalších 26 mutací s reziduální funkcí, u nichž byla prokázána odpovídavost k tezakaftoru v kombinaci s ivakaftorem [11]. Novou generaci CFTR korektoru představuje elexakaftor. V kombinaci s tezakaftorem a ivakaftorem (KaftrioTM, Vertex Pharmaceuticals) představuje průlomový lék pro pacienty nesoucí mutaci F508del na obou alelách nebo v heterozygotní konstituci s mutací spojenou s minimální funkcí. Schválen ke klinickému použití byl v roce 2019, klinické studie prokázaly významné zlepšení funkce plic, snížení hodnoty chloridů v potu a zlepšení stavu výživy [12].
Přehled modulátorů CFTR proteinu
schválených ke klinickému použití Evropskou lékovou
agenturou (European Medicines Agency, EMA) uvádí tabulka 1
[13‒16].
Závěr
Dlouhodobě představovaly všechny možnosti terapie CF léčbu symptomatickou: fyzioterapie, mukolytika, antibiotika, léky s protizánětlivým účinkem a léky vedoucí ke zlepšení stavu výživy pacienta. Snaha o další zlepšení v rámci těchto standardních terapií postupuje v posledních několika letech paralelně s mohutným rozvojem kauzální léčby CF. Jejím cílem je ovlivnit molekulární defekt jako příčinu samotného onemocnění. V současné době jsou k dispozici čtyři CFTR modulátory, které dohromady mohou účinkovat až u 90 % pacientů s CF. Specifická kauzální léčba se postupně stává součástí standardní terapie pacientů s tímto život limitujícím onemocněním.
Seznam použité literatury
- [1] Sosnay PR, Siklosi KR, Van Goor F, et al. Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nat Genet 2013; 45: 1160–1167.
- [2] Cutting GR. Cystic fibrosis genetics: From molecular understanding to clinical application. Nat Rev Genet 2015; 16: 45–56.
- [3] Boeck K De, Amaral MD. Rapid Review Progress in therapies for cystic fibrosis. Lancet Respir Med 2016; 2006: 1–13.
- [4] Riordan JR, Rommens JM, Kerem BS, et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989; 245: 1066–1073.
- [5] CFTR2. Resources; CFTR2 2017. Dostupné na: https://cftr2.org/resources
- [6] Bobadilla JL, Macek M, Fine JP, Farrell PM. Cystic fibrosis: A worldwide analysis of CFTR mutations ‒ Correlation with incidence data and application to screening. Hum Mutat 2002; 19: 575–606.
- [7] Ramsey BW, Davies J, McElvaney NG, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med 2011; 365: 1663–1672.
- [8] Wainwright CE, Elborn JS, Ramsey BW, et al. Lumacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med 2015; 373: 220–231.
- [9] Taylor‑Cousar JL, Munck A, McKone EF, et al. Tezacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N Engl J Med 2017; 377: 2013‒2023.
- [10] Guimbellot J, Solomon GM, Baines A, et al. Effectiveness of ivacaftor in cystic fibrosis patients with non‑G551D gating mutations. J Cyst Fibros 2019; 18: 102–109.
- [11] Rowe SM, Daines C, Ringshausen FC, et al. Tezacaftor–ivacaftor in residual‑function heterozygotes with cystic fibrosis. N Engl J Med 2017; 377: 2024–2035.
- [12] Middleton PG, Mall MA, Dřevínek P, et al. Elexacaftor‑tezacaftor‑ivacaftor for cystic fibrosis with a single Phe508del allele. N Engl J Med 2019; 381: 1809–1819.
- [13] SPC Kalydeco, European Medicines Agency. Dostupné na: https://www.ema.europa.eu/en/medicines/human/EPAR/kalydeco [accessed September 26, 2020]
- [14] SPC Orkambi, European Medicines Agency. Dostupné na: https://www.ema.europa.eu/en/medicines/human/EPAR/orkambi [accessed September 26, 2020]
- [15] SPC Symkevi, European Medicines Agency. Dostupné na: https://www.ema.europa.eu/en/medicines/human/EPAR/symkevi [accessed September 26, 2020]
- [16] SPC Kaftrio, European Medicines Agency. Dostupné na: https://www.ema.europa.eu/en/medicines/human/EPAR/kaftrio [accessed September 26, 2020]