Pirfenidon v léčbě idiopatické plicní fibrózy
Souhrn:
Idiopatická plicní fibróza (IPF) je charakterizována difuzním procesem primární fibroproliferace v plicní tkáni s radiologickým a histopatologickým obrazem běžné intersticiální pneumonie. Jde o závažné onemocnění, které bez léčby nevyhnutelně vede k úmrtí pacienta. V Evropě se počet pacientů s IPF odhaduje na přibližně 110 000, je však možné, že počet nemocných je ještě vyšší vzhledem k nedostatečné diagnostice této nemoci. Střední doba přežití pacientů s IPF bez léčby je 2,5–3 roky. Do nedávné doby nebyl pro terapii IPF dostupný žádný účinný lék. Od roku 2011 je však v Evropě již dostupný první přípravek, který ovlivňuje hojení fibroproliferativních změn v plicní tkáni u IPF, a to pirfenidon. Tato látka dokáže zpomalit, nebo dokonce zastavit progresi fibrózních změn v plicích a tak u pacientů s IPF zpomalit pokles plicních funkcí. Tím dochází ke zlepšení jejich prognózy a ke snížení mortality na tuto závažnou nemoc. Toto léčivo je v ČR od 1. 7. 2014 hrazeno ze systému veřejného zdravotního pojištění pacientům s mírnou až středně těžkou IPF a je dostupné v Centrech pro diagnostiku a léčbu intersticiálních plicních procesů, jejichž seznam lze najít na stránkách České pneumologické a ftizeologické společnosti ČLS JEP (www.pneumologie.cz). Každý pacient s podezřením na IPF by tedy měl být co nejdříve odeslán do některého z center, aby byl včas vyšetřen a v případě potvrzené diagnózy včas léčen specifickou antifibrotickou léčbou. Jedině tak je možné těmto pacientům prodloužit život a zlepšit jeho kvalitu.
Key words: idiopathic pulmonary fibrosis, pirfenidone, indications, reimbursement rules.
Summary:
Idiopathic pulmonary fibrosis (IPF) is a diffuse, primarily fibroproliferative disorder of lungs characterized by radiologic and histopathologic pattern of usual interstitial pneumonia. IPF is a severe disease which – if untreated – leads inevitably to death. It is estimated that about 110.000 patients with this disease live in Europe but the count might be even higher due to undiagnosed cases. Median survival of the patients with untreated IPF is 2.5 3 years. Until very recently, there was no effective drug against this disease. The first drug which directly affects fibroproliferative healing in IPF, pirfenidone, is available in Europe since 2011. This drug can slow down or stop progression of lung fibrosis and thus slow down decline of lung functions as well. As a result, the prognosis of IPF patients is improved and mortality decreased. Pirfenidone is reimbursed in the Czech Republic from general health insurance funds since 1. 7. 2014 for the patients with mild or moderate IPF and is available in the Centers for diagnosis and treatment of interstitial lung diseases recognized by Czech Pneumologic and Phtiseologic Society (see www.pneumologie.cz). Every patient with suspected IPF should be sent to some of these centres as early as possible, to be immediately checked and in case of IPF confirmation treated early by specific antifibrotic treatment. This is the only chance for IPF patients to prolong their survival and to improve the quality of their lives.
Úvod
Idiopatická plicní fibróza (IPF) je definována jako specifická forma chronického fibrotizujícího intersticiálního plicního procesu dospě-lých nejasné etiologie s histologickým a radiologickým obrazem běžné intersticiální pneumonie. Vyskytuje se výhradně u dospělých, a to středního a staršího věku. Poslední konsensus světových respiračních společností o diagnóze a léčbě IPF z roku 2011 změnil pohled na toto onemocnění, a to na základě medicíny založené na důkazech [1]. Zásadním přínosem tohoto dokumentu je důrazné nedoporučení léčby kortikosteroidy a imunosupresivy, která pacientům s IPF přinášela většinou pouze obtíže z důvodu nežádoucích účinků bez jaké-hokoli léčebného přínosu. Navíc se IPF poprvé stává nemocí potenciálně léčitelnou, a to díky objevu prvního antifibroticky působícího léčiva – pirfenidonu.
Epidemiologie IPF
Epidemiologie IPF není přesně známa, ale výskyt onemocnění zřejmě stoupá. V devadesátých letech minulého století dosahovala inci-dence kolem 3/100 000 obyvatel, nyní americké registry odhadují prevalenci 13–20/100 000 obyvatel a incidenci 7,4/100 000 u žen a 10,7/100 000 u mužů. Pacienti s IPF jsou nejčastěji ve středním věku, v rozmezí 40–70 let. Idiopatická plicní fibróza nemá podle pro-vedených výzkumů žádnou jednoznačnou geografickou distribuci, vyskytuje se celosvětově s přibližně stejnou prevalencí, bez rozdílů mezi městským a venkovským obyvatelstvem a bez jakékoli spojitosti s rasou nebo etnicitou. Objevuje se obvykle sporadicky, familiární výskyt je vzácný [2].
Etiopatogeneze IPF
V poznání etiopatogeneze IPF byl v posledních letech učiněn významný pokrok. Předpokládá se, že jde o patologický vzorec hojení opakovaných alveolárních lézí různé etiologie. V důsledku alveolárního poranění dochází k apoptóze buněk epiteliální výstelky, je obna-žena bazální membrána, a tím spuštěna kaskáda aktivace cytokinů, chemokinů a enzymů, které se pak podílejí na dalším rozvoji a udržování procesu fibroproliferace a jizvení plicní tkáně [3]. V tomto procesu hraje nepochybně roli nerovnováha oxidačních a enzymatických pochodů a nerovnováha spektra cytokinů. Oxidační stres se v patogenezi IPF uplatňuje především při vzniku iniciálních mikroskopických lézí alveolárního epitelu a podílí se na nemožnosti úspěšného zhojení těchto lézí; tím, že přímo potencují fibroprolife-raci cestou transformujícího růstového faktoru beta (transforming growth factor beta, TGFβ), a navíc myofibroblasty diferencované vlivem TGFβ jsou samy zdrojem reaktivních kyslíkových radikálů. V rámci nerovnováhy proteolytických enzymů v patogenezi IPF je zřejmě dominantní nepoměr spektra metaloproteáz a jejich tkáňových inhibitorů v intracelulárním a extracelulárním kompartmentu.
Z cytokinů hrají v etiopatogenezi IPF pravděpodobně zásadní roli cytokiny Th2, a to interleukiny 4 a 13 (IL 4, IL 13). Oba podporují růst fibroblastů a produkci kolagenu a zvyšují produkci TGFβ2 lidskými bronchiálními epiteliemi. TGFβ je klíčovým cytokinem v indukci fibroproliferativního hojení alveolárního poranění. Indukuje transkripci genu kolagenu typu I a fibronektinu ve fibroblastech. Výskyt fibroblastů v plicní tkáni, kde za normálních okolností nejsou přítomny, má několik možných zdrojů a mechanismů. Jednak fibroblasty vznikají z rezidentních buněk, které se aktivují a proliferují jako odpověď na poranění. Je možný i jejich původ z alveolárních epiteliálních buněk, které projdou tzv. epitelo mezenchymální transdiferenciací. Část fibroblastů je však pravděpodobně mimoplicního původu, a to z mezenchymálních kmenových buněk kostní dřeně, které jsou atrahovány do míst poranění v plicní tkáni. Je pravděpodob-né, že fibroblasty u jedinců s IPF mají i sníženou schopnost apoptózy a že jsou přímo chráněny proti apoptóze zprostředkované Fas inhibitory apoptózy (inhibitor apoptosis protein, IAP) [4].
Na vznik IPF má nepochybně vliv i senescence, neboť se s nemocí nesetkáme u jedinců mladších než 45 let. Působí zde také genetic-ká výbava jedince, ta určuje genetickými mutacemi a variacemi (polymorfismy) jak složení a množství mucinu v plicích (polymorfismy genu pro mucin – MUC 5B), tak složení cytokinového spektra (polymorfismy genu pro IL 4 mohou kupříkladu ovlivnit produkci tohoto cytokinu) [5]. Navíc u familiárně vázaných IPF mohou být přítomny mutace genů pro telomerázy, které přímo ovlivňují sklon k časné senescenci, či mutace genů pro surfaktant, jež jsou pak často příčinou dětských forem plicních fibróz.
Klinický obraz IPF
Idiopatická plicní fibróza se klinicky projevuje progredující námahovou a posléze klidovou dušností, snadnou unavitelností, kašlem a v pozdějších fázích při nastupující hypoxemii i cyanózou. U tří čtvrtin pacientů se také vyskytují fenotypové projevy, jako jsou palič-kovité prsty s nehty tvaru hodinového sklíčka a poslechový fenomén krepitu slyšitelný nad plicními bazemi. Přestože je pro IPF typický pozvolný a plíživý nástup dušnosti s pomalu progredujícím zhoršováním, u některých pacientů se vyskytnou epizody tzv. akutní exacer-bace IPF, kdy dojde k náhlému klinickému zhoršení s poklesem plicních funkcí a radiologickým obrazem tzv. opacit mléčného skla svědčícím pro akutní difuzní poškození alveolů [6].
Pro diagnózu IPF je zcela zásadní radiologický a histopatologický obraz běžné intersticiální pneumonie (usual interstitial pneumonia, UIP). V radiologickém obraze získaném metodou výpočetní tomografie hrudníku s vysokým rozlišením (high resolution computed to-mography, HRCT) je obraz UIP charakterizován retikulárními opacitami s obrazem voštinovité plíce s maximem subpleurálně a v bazích plicních a minimálním rozsahem okrsků charakteru mléčného skla, které charakterizují alveolární a zánětlivé změny. Histopatologický obraz je u IPF různorodý podle lokalizace odběru plicní tkáně, většina odebraných vzorků však zachycuje obraz UIP s převahou jizvení a s destrukcí plicní architektoniky [7].
Funkčně je IPF charakterizována restrikční ventilační poruchou a významnou poruchou plicní poddajnosti. Hodnota transfer faktoru (TLCO) je významně snížena, a to ve větší míře, než by odpovídalo redukci vitální kapacity (vital capacity, VC). Charakteristickým nále-zem u IPF je hypoxemie, zprvu pouze námahová, posléze i klidová. Funkční vyšetření hraje významnou roli při sledování nemocných s IPF. Výchozí hodnota VC a TLCO a jejich pokles v čase totiž určují prognózu nemocných s IPF a také indikaci antifibrotické léčby a její účinnost.
Bronchoalveolární laváž (BAL) dříve patřila mezi vyšetřovací metody, kterým se v diagnostice a sledování IPF přikládala velká důležitost. V současnosti má význam spíše z hlediska diferenciální diagnostiky. Pro IPF je typické zmnožení neutrofilních granulocy-tů v tekutině získané BAL, obvykle s malou příměsí eosinofilů, počty lymfocytů bývají zvýšeny minimálně. U pacientů s typickým klinickým obrazem odpovídajícím IPF a typickým radiologickým nálezem na HRCT hrudníku obvykle není plicní biopsie indiková-na. Pokud však klinický či radiologický obraz není typický, pak pro utvrzení diagnózy vzorek plicní tkáně potřebujeme. Nově máme možnost volby mezi méně zatěžující bronchoskopickou transbronchiální biopsií metodou kryobiopsie se získáním vzorků až o průměru okolo 1 cm a chirurgickou plicní biopsií, která je stále zlatým standardem pro histopatologickou diagnostiku IPF [6,8].
Léčba IPF
Průlomem v přístupu k diagnostice a hlavně léčbě IPF byl konsensus světových společností zabývajících se respiračními onemocněními z roku 2011. Při tvorbě tohoto konsensu byly provedeny metaanalýzy validních studií z posledních deseti let a vyhodnoceny léčebné postupy užívané pro léčbu IPF do roku 2009. Potvrdil se očekávaný závěr, že u IPF prakticky vůbec neúčinkují protizánětlivě působící léčiva, jako jsou kortikosteroidy, imunosupresiva a cytostatika, a tudíž by se neměla nadále používat v její léčbě, a to ani samotná, ani v kombinacích [1]. Tento závěr byl ještě podpořen interim analýzami studie PANTHER zveřejněnými na jaře 2012, na jejichž základě bylo pozastaveno pokračování studijní větve pacientů s IPF léčených kombinací prednison + azathioprin + N acetylcystein (NAC), neboť zde byla zjištěna vyšší mortalita oproti větvi se samotným NAC i ve srovnání s větví, v níž bylo podáváno placebo [9].
Převrat v léčbě IPF znamenal zcela jednoznačně objev antifibrotického účinku malé molekuly – pirfenidonu – a následně registrace tohoto přípravku a posléze i úhrada, nejprve v Japonsku, následně v Evropě a nyní i v Kanadě a USA.
Pirfenidon v léčbě IPF
Mechanismus účinku pirfenidonu
Pirfenidon je prvním léčivem, které zasahuje přímo do patogeneze IPF, i když mechanismus jeho účinku není doposud v detailech objas-něn. Nicméně existují důkazy pro antifibrotické i protizánětlivé účinky tohoto léčiva v modelu bleomycinem indukované a posttransplantační plicní fibrózy. Pirfenidon snižuje proliferaci fibroblastů a produkci proteinů a cytokinů spojených s fibrózou pravdě-podobně inhibicí TGFβ1 a destičkového růstového faktoru (platelet derived growth factor, PDGF) cestou blokády nukleární translokace proteinu SMAD (Sma and Mad related protein).
Klinická účinnost léčby pirfenidonem
Klinická účinnost léčby pirfenidonem byla prokázána v multinárodní studii CAPACITY 004 (PIPF 004) a v japonské studii Shionogi (SP3). Nicméně účinek léčby pirfenidonem na zmírnění poklesu plicních funkcí již nebyl statisticky signifikantní ve studii CAPACITY 006 (PIPF 006). Ve společné analýze CAPACITY 004 a 006 však byl prokázán vliv léčby pirfenidonem na pokles mortality ve srovnání s placebem (7,8 % ve skupině s pirfenidonem vs. 9,8 % ve skupině s placebem) [10].
Studie ASCEND (PIPF 016) byla následně navržena a provedena s cílem potvrdit pozitivní účinek pirfenidonu na zpomalení progre-se IPF, prokázaný dvěma předchozími studiemi. Studie se účastnilo 555 pacientů s IPF a byla kontrolována placebem. Primárním cílo-vým ukazatelem byla změna VC nebo úmrtí v 52. týdnu. Sekundárním cílovým ukazatelem byla změna vzdálenosti překonané za 6 minut (6 MWD, 6 minute walk distance), čas do progrese nemoci (progression free survival, PFS), dušnost a úmrtí ze všech příčin. Ve skupině léčené pirfenidonem bylo v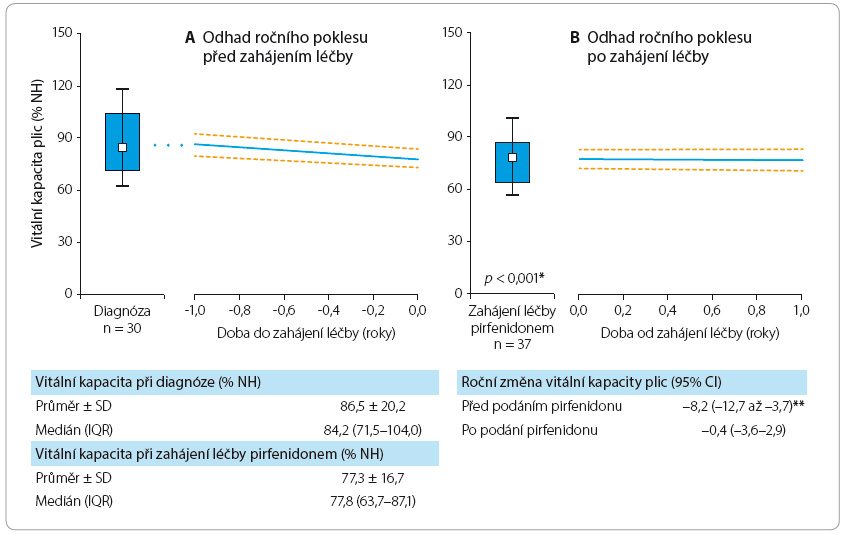 porovnání s placebem o 47,9 % méně pacientů, u nichž nastal pokles VC o 10 % a více oproti výchozí hodnotě nebo kteří zemřeli. Rovněž byl v této skupině patrný vyšší počet pacientů (13,2 %), u nichž při léčbě pirfenidonem ve srovnání s pacienty ze skupiny s placebem nedošlo k žádnému poklesu VC; pirfenidon navíc statisticky významně snižoval i pokles 6 MWD a prodlužoval PFS. Společnou analýzou studií CAPACITY 004 a 006 a ASCEND bylo navíc prokázáno snížení mortality ze všech příčin i na IPF ve skupině léčené pirfenidonem oproti placebu.
porovnání s placebem o 47,9 % méně pacientů, u nichž nastal pokles VC o 10 % a více oproti výchozí hodnotě nebo kteří zemřeli. Rovněž byl v této skupině patrný vyšší počet pacientů (13,2 %), u nichž při léčbě pirfenidonem ve srovnání s pacienty ze skupiny s placebem nedošlo k žádnému poklesu VC; pirfenidon navíc statisticky významně snižoval i pokles 6 MWD a prodlužoval PFS. Společnou analýzou studií CAPACITY 004 a 006 a ASCEND bylo navíc prokázáno snížení mortality ze všech příčin i na IPF ve skupině léčené pirfenidonem oproti placebu.
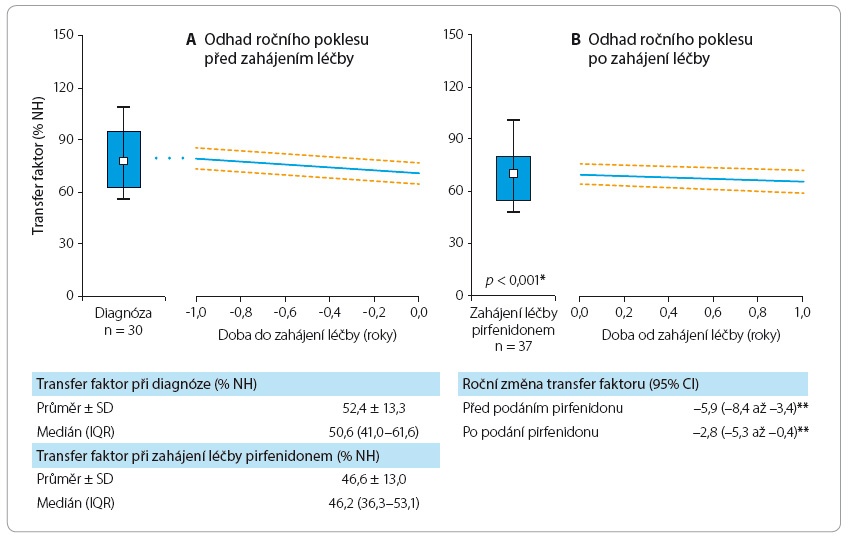
Pirfenidon tedy jednoznačně prokázal léčebný efekt u pacientů s IPF, a byl dokonce označen americkým úřadem Food and Drug Administration jako průlomový lék v terapii dosud neléčitelné nemoci – IPF [11]. Účinnost léčby IPF pirfenidonem byla prokázána i u naší populace léčených pacientů, a to na základě statistického zpracování dat z Registru idiopatické plicní fibrózy České pneumolo-gické a ftizeologické společnosti (ČPFS) spravovaného Institutem biostatistiky a analýz Lékařské a Přírodovědecké fakulty Masarykovy univerzity Brno (graf 1a, b, graf 2a, b).
Dostupnost a úhrada pirfenidonu v ČR
V ČR byl pirfenidon dostupný již před ukončením studie ASCEND (PIPF 016), a to v rámci tzv. časného přístupu v programu na jméno pacienta (Named Patient Programme, NPP). Po ukončení NPP v roce 2012, z důvodu registrace pirfenidonu v Evropské unii, byl přípravek dovážen do Čech pro pacienty s IPF mechanismem mimořádného dovozu s úhradou na základě paragrafu 16 zákona o veřejném zdravotním pojištění (48/1997 Sb). I v tomto přechodném období tedy měli pacienti s IPF v ČR přístup k cílené léčbě své nemoci. Cena a úhrada přípravku je v ČR stanovena od 1. 7. 2014. Pirfenidon je dostupný v režimu vysoce inovativního léčivého pří-pravku (VILP) pro pacienty s IPF s VC v rozmezí 50–80 % normálních hodnot (NH) a s TLCO vyšším než 35 % NH, a to v Centrech pro diagnostiku a léčbu intersticiálních plicních procesů (seznam na www.pneumologie.cz), navíc po předchozím souhlasu revizního lékaře s léčbou (preskripční omezení W). Pacienti musejí splnit výše uvedená vstupní kritéria pro léčbu pirfenidonem, nesmějí kouřit, a navíc u nich musí být prokázána i účinnost léčby. V praxi to znamená, že jsou pravidelně nejméně po šesti měsících kontrolováni a při kontrolách nesmí být patrný pokles VC ≥ 10 % a pokles hodnot TLCO ≥ 15 % ve třech po sobě následujících šestiměsíčních inter-valech. Každý pacient léčený pirfenidonem také musí být zaznamenán v Registru VILP, který je podregistrem Registru pacientů s idiopatickou plicní fibrózou.
Dávkování pirfenidonu v léčbě IPF
Pacienti zahajují léčbu 1.–7. den dávkou 3krát 1 kapsle (cps) pirfenidonu po 267 mg, 8.–15. den se pak dávka zvyšuje na 3krát 2 cps po 267 mg a od 16. dne pak pacienti užívají 3krát 3 cps po 267 mg, čímž dosáhnou cílové dávky 2 403 mg/den. Pacienti, kteří kdykoli v průběhu užívání léčbu přeruší na více než dva týdny, začínají opět iniciální fází léčby. U pacientů, kteří trpí výraznými nežádoucími gastrointestinálními účinky (viz níže), může být denní dávka pirfenidonu snížena na nejvyšší tolerovanou (kupříkladu 3krát 2 cps nebo pouze 3krát 1 cps po 267 mg). Také v případě fotosenzitivní reakce či významné elevace hodnot jaterních testů je zpravidla nutné pře-chodné snížení dávky či úplné přerušení léčby (viz níže).
Poučení pacienta při zahájení léčby pirfenidonem
Každý pacient musí být při zahájení léčby pirfenidonem poučen o nutnosti zanechání kouření, jinak by byla účinnost léčby ohrožena (viz níže). Musí být seznámen s fototoxicitou léčiva a s nutností soustavné ochrany proti slunečnímu záření (celoročně krémy s vysokým ochranným faktorem, tzv. sun blockery, na nechráněná místa kůže a pokrývka hlavy a ochrana těla oděvem při riziku oslunění). Pacient také musí vědět o gastrointestinálních nežádoucích účincích, zejména o nevolnosti a nechuti k jídlu, a o možnosti snížit dávku při jejich výskytu. Musí být poučen i o nutnosti opakovaného laboratorního vyšetření krve v průběhu léčby pirfenidonem z důvodu rizika poruchy funkce jater.
Kontraindikace podání pirfenidonu
Léčba pirfenidonem je kontraindikována při alergii na pirfenidon, při výskytu angioedému po podání pirfenidonu v anamnéze, při sou-časném užívání fluvoxaminu, trpí li pacient závažnou poruchou jaterních funkcí nebo jaterním onemocněním v konečné fázi a při závaž-ném onemocnění ledvin (clearance kreatininu < 30 ml/min) nebo při renálním selhání vyžadujícím dialýzu.
Nežádoucí účinky pirfenidonu a jejich zvládání
Gastrointestinální obtíže a intolerance
Trávicí obtíže, jako je nauzea, nechutenství, případně až úbytek hmotnosti, jsou poměrně časté (až jedna třetina pacientů léčených pirfe-nidonem), dají se však obvykle zvládnout podáváním přípravku spolu s jídlem, někdy je zapotřebí snížit dávku na nejvyšší tolerovanou.
Fotosenzitivita
Jedním z relativně častých nežádoucích účinků je fotosenzitivita a kožní vyrážky po oslunění (přibližně jedna čtvrtina pacientů léčených pirfenidonem). Proto musí být nemocní náležitě poučeni, aby se chránili před osluněním, a to pokud možno zakrytím většiny těla oděvem a ošetřením exponované části kůže krémy s vysokým ochranným faktorem. Málokdy je kožní vyrážka závažná a vyžaduje snížení dávky (obvykle na dávku 3krát denně 1 cps po dobu 7 dnů), případně úplné přerušení léčby pirfenidonem do ústupu kožních změn.
Porucha jaterních funkcí
U pacientů léčených pirfenidonem, a to i bez anamnézy předchozího onemocnění jater, je popisována poměrně často izolovaná elevace hodnot aminotransferáz (aspartátaminotransferázy, AST, a alaninaminotransferázy, ALT) na více než trojnásobek horního limitu normy. Proto je nutné absolvovat vstupní laboratorní vyšetření aminotransferáz, které je pak vhodné opakovat v měsíčních intervalech v prvních šesti měsících a dále při každé klinické kontrole IPF. V případě přetrvávající zvýšené aktivity aminotransferáz je nutná úprava dávky pirfenidonu či přechodné přerušení léčby. Zároveň je potřeba pokud možno eliminovat další vlivy, které se mohou podílet na zvýšení aktivity aminotransferáz (alkohol, léky). V případě elevace hodnot aminotransferáz na méně než pětinásobek normy, avšak s elevací hodnot bilirubinu, či v případě elevace aminotransferáz nad pětinásobek horní hranice normy je nutné léčbu pirfenidonem ukončit. U pacientů s onemocněním jater (Child Pugh A, B) je třeba věnovat vyšší pozornost monitoraci aminotransferáz vzhledem k dosažení vyšších koncentrací pirfenidonu (až o 60 %), zvláště pokud zároveň užívají inhibitor CYP1A2 (viz níže). U pacientů s těžkým poškoze-ním jater by neměl být pirfenidon podáván.
Angioedém
Angioedém není častou komplikací podávání pirfenidonu a byl popsán až v postmarketingovém sledování. Objevuje se většinou v oblasti hlavy a krku spolu s otoky sliznic a jazyka s pískoty a dušností. U těchto pacientů musí být léčba pirfenidonem okamžitě ukončena a lék by neměl být již nikdy podán.
Další nežádoucí účinky
Závratě, točení hlavy a únava nejsou častou komplikací léčby pirfenidonem a obvykle vymizí spontánně bez nutnosti přerušení léčby či redukce dávky léčiva.
Interakce pirfenidonu
Pirfenidon je metabolizován převážně cestou CYP1A2. Vzhledem k tomu současné podávání látek a léčiv omezujících či blokujících tento enzym ovlivňuje výrazně metabolismus pirfenidonu.
Inhibitory CYP1A2
Mezi přírodní inhibitory CYP1A2 patří grapefruitová šťáva, neměla by proto být v průběhu léčby pirfenidonem konzumována. Z léčiv je významným inhibitorem fluvoxamin a jeho užívání je kontraindikací k zahájení léčby pirfenidonem; podávání fluvoxaminu musí proto být před léčbou pirfenidonem přerušeno. Enoxacin inhibuje i CYP1A2, čímž 2–4krát zvyšuje expozici pirfenidonu. Pokud však není možné se podávání tohoto léčiva vyhnout, je třeba snížit denní dávku pirfenidonu na 3krát 1 cps po 267 mg. V případě současného podá-vání ciprofloxacinu je předpokládaná dostupnost pirfenidonu zvýšena o 81 % a jeho denní dávka by tedy měla být snížena na 3krát 2 cps po 267 mg. U pacientů užívajících pouze mírné inhibitory CYP1A2 (amiodaron, propafenon) pouze pečlivěji kontrolujeme výskyt nežá-doucích účinků a jaterní funkce.
Induktory CYP1A2
Vzhledem k tomu, že tabákový kouř je induktorem CYP1A2, ovlivňuje významně farmakokinetiku a tím biologickou dostupnost (expo-zici) pirfenidonu, a to až o 50 %, a proto by pacienti neměli kouřit. Navíc nekuřáctví je samo o sobě jednou z podmínek úhrady pirfeni-donu v ČR.
Významným induktorem CYP1A2 je též jedno z nejúčinnějších antituberkulotik – rifampicin. Současnému podávání pirfenidonu a rifampicinu bychom se proto měli pokud možno vyhnout.
Inhibitory protonové pumpy (jmenovitě omeprazol) jsou také slabými induktory CYP1A2. Vzhledem k tomu, že u pacientů s IPF po-dáváme tyto přípravky jako prevenci vzniku alveolárních poranění na podkladě asymptomatického refluxu, hrozí snížení biologické dostupnosti současně podávaného pirfenidonu. Výhodnější se v tomto případě jeví podávání esomeprazolu, který indukuje CYP1A2 podstatně méně.
Další možnosti léčby IPF
Jiná antifibrotická léčiva
Do 15. 1. 2015 byl v tzv. časném přístupu pro pacienty s IPF dostupný nintedanib (trikinázový inhibitor), který v dávce 150 mg podávané 2krát denně perorálně také prokazatelně snižuje pokles plicních funkcí [12,13]. Bylo možné jej podat pacientům, kteří nesplnili podmín-ky úhrady pro léčbu pirfenidonem, tj. měli VC vyšší než 80 %, případně TLCO v rozmezí 30–35 % NH nebo netolerovali pirfenidon, případně na něj byli alergičtí či se u nich vyskytly závažné nežádoucí účinky léčby. Od 15. 1. 2015 je nintedanib registrován v Evropě pro diagnózu IPF a čeká na stanovení ceny a úhrady. Skončila tudíž možnost zařazování pacientů do specifického léčebného programu.
Jiná užívaná farmakoterapie
U pacientů s IPF obvykle podáváme ještě NAC v dávce 600 mg 3krát denně, i když nebyl prokázán jeho vliv na zabránění poklesu plic-ních funkcí, a to pro jeho antioxidační účinek, s cílem předcházet dalšímu poškozování alveolárního epitelu oxidačními ději, a tím bránit tzv. akutním exacerbacím. N acetylcystein je dobře tolerován, některým pacientům vadí pouze zvýšená expektorace [6,14]. Od 1. 6. 2013 je NAC v ČR hrazen pro pacienty s IPF z prostředků veřejného zdravotního pojištění.
Z důvodu prevence akutní exacerbace IPF podáváme všem pacientům také inhibitory protonové pumpy (nejlépe esomeprazol, viz výše) v běžných denních dávkách, navzdory tomu, že jsme si vědomi jeho možných interakcí s pirfenidonem prostřednictvím CYP1A2. Bezpříznakový extraezofageální reflux může být také příčinou nových alveolárních lézí, a proto i spouštěčem akutních exacerbací.
Dušnost u pacientů v terminálních stadiích IPF obvykle tlumíme opiáty, a to většinou v náplasťových formách s kontinuálním uvolňováním.
Klinické studie
V současné době probíhají další klinické studie zkoušející léčiva, která by měla přímo zasahovat do patogenetických pochodů IPF. Paci-enty s IPF, obzvláště ty, kteří nesplní kritéria pro léčbu pirfenidonem, je vhodné pokud možno do těchto studií zařazovat. Ze zkoušených potenciálních nových přípravků pro léčbu IPF je vhodné zmínit monoklonální protilátku proti IL 13 lebrikizumab a inhibitor molekuly lysyl oxidase like anti LOXL 2.
Akutní exacerbace IPF
Při akutní exacerbaci IPF podáváme pacientům vysoké dávky kortikosteroidů parenterálně (obvykle 0,5–1 g methylprednisolonu i.v. po dobu tří dnů s následným snížením na 1 mg/kg i.v.) a při podezření na současnou infekci jako spouštěč podáváme také antibiotika. Při respirační insuficienci v průběhu akutní exacerbace ústící až v respirační selhání se nekloníme k umělé plicní ventilaci, neboť pro pacien-ty většinou není žádným přínosem, dochází při ní k ještě rozsáhlejšímu poškození plicní tkáně s následným úmrtím pacienta. Umělou plicní ventilaci bychom tedy volili pouze u těch pacientů s akutní exacerbací, kteří jsou již zařazeni na čekací listinu transplantace plic, a mají tedy alespoň nějakou šanci se darované plíce dočkat.
Dlouhodobá domácí oxygenoterapie
V případě pokročilého onemocnění s hypoxemií indikujeme pacientům, kteří splní kritéria podle ČPFS, dlouhodobou domácí oxygenote-rapii, a to buď koncentrátorem kyslíku, nebo kyslíkem kapalným, který je indikován pro pacienty vyžadující vysoké průtoky kyslíku a pro pacienty mobilní, kdy umožňuje rehabilitaci i pohyb mimo domácí prostředí s předplněnou lahví kapalného kyslíku.
Transplantace plic
Pro některé přísně vybrané pacienty je vhodná transplantace plic, a to většinou jedné plíce, vzácně bloková transplantace srdce a plic. Kritérii pro zařazení na čekací listinu jsou v době diagnózy: dušnost, TLCO < 40 % NH, saturace < 88 % PaO2 (parciální tlak kyslíku v tepenné krvi) a obraz voštinovité plíce na HRCT. V případě longitudinálního sledování je indikací ke zvážení transplantace plic pokles usilovné vitální kapacity (forced vital capacity, FVC) o 10 % a pokles TLCO o 15 % spolu se zhoršením fibrózy zjištěné při HRCT hrudníku.
Rehabilitace
Jednou z možností, jak zlepšit kvalitu života pacienta, je zlepšení jeho funkční výkonnosti a zmírnění dušnosti. Rehabilitace musí být v případě onemocnění IPF komplexní, zahrnující učení, konzultace a behaviorální techniky ke zlepšení sebeobsluhy, dále redukci sym-ptomů a optimalizaci funkční kapacity.
Závěr
Idiopatická plicní fibróza je nepochybně velmi závažné onemocnění, jehož časnému záchytu, diagnostice a časné cílené léčbě bychom určitě měli věnovat pozornost. S pacienty s IPF bychom měli zacházet stejně jako s onkologicky nemocnými, jejich onemocnění je bez léčby také fatální a je léčitelné pouze časným zahájením specifické cílené léčby. Pirfenidon je nepochybně průlomovým léčivem, prvním v léčbě IPF, které změnilo toto onemocnění z doposud neléčitelného na potenciálně léčitelné.
Práce byla podpořena grantem NT 13433 4/2012, IGA MZČR.
Seznam použité literatury
- [1] Raghu G, Collard HR, Egan JJ, et al. ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence based guideli-nes for diagnosis and management. Am J Respir Crit Care Med 2011; 183: 788–824.
- [2] Nalysnyk L, Cid Ruzafa J, Rotella P, Esser D. Incidence and prevalence of idiopathic pulmonary fibrosis: review of the literature. Eur Respir Rev 2012; 12: 355–361.
- [3] Loomis King H, Flaherty KR, Moore BB. Pathogenesis, current treatments and future directions for idiopathic pulmonary fibrosis. Curr Opin Pharmacol 2013; 3: 377–385.
- [4] Borensztajn K, Crestani B, Kolb M. Idiopathic pulmonary fibrosis: from epithelial injury to biomarkers insights from the bench side. Respiration 2013; 6: 441–452.
- [5] Vasakova M, Striz I, Slavcev A, et al. Th1/Th2 cytokine gene polymorphisms in patients with idiopathic pulmonary fibrosis. Tissue Antigens 2006; 3: 229–232.
- [6] Kolek V, Kašák V, Vašáková M. Pneumologie. 2. vydání, Praha: Maxdorf, 2014. s. 261–264.
- [7] Smith M, Daluzro M, Panse P, et al. Usual interstitial pneumonia pattern fibrosis in surgical lung biopsies. Clinical, radiological and histopathological clues to aetiology. J Clin Pathol 2013; 10: 896–903.
- [8] Wells AU. Managing diagnostic procedures in idiopathic pulmonary fibrosis. Eur Respir Rev 2013; 128: 158–162.
- [9] McGrath EE, Millar AB. Hot off the breath: triple therapy for idiopathic pulmonary fibrosis hear the PANTHER roar. Thorax 2012; 2: 97–98.
- [10] Noble PW, Albera C, Bradford WZ, et al. CAPACITY Study Group. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet 2011; 377: 1760–1769.
- [11] King TE Jr, Bradforf WB, Catsro Bernardini S, et al.; ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 22: 2083–2092.
- [12] Richeldi L, Du Bois RM, Raghu G, et al.; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 22: 2071–2082.
- [13] Antoniou KM, Margaritopoulos GA, Siafakis NM. Pharmacological treatment of idiopathic pulmonary fibrosis: from the past to the future. Eur Respir Rev 2013; 129: 281–291.
- [14] Vašáková M a kol. Moderní farmakoterapie v pneumologii. Praha: Maxdorf, 2013. s. 236–243.