Rekombinantní přípravky třetí generace v léčbě hemofilie
Souhrn:
Léčba hemofilie je v dnešní době účinná a bezpečná. Kvalita života pacientů se za posledních dvacet let nesrovnatelně zlepšila. S nástupem rekombinantních derivátů třetí generace, které jsou vyráběny technologiemi rekombinantní DNA bez přidání zvířecí či lidské plazmatické bílkoviny v kterékoliv fázi výroby, došlo k eliminaci jakékoliv krví přenosné nákazy během léčby. Nejnovější možností léčby jsou deriváty s prodlouženým účinkem, jejichž biologický poločas eliminace je zhruba 1,5× delší než biologický poločas eliminace ostatních rekombinantních derivátů pro léčbu hemofilie A a 3–6× delší než biologický poločas eliminace derivátů pro léčbu hemofilie B. Současnou nejzávažnější komplikací léčby hemofilie je vznik inhibitoru – protilátek proti podávanému derivátu.
Key words: hemophilia A – hemophilia B – third generation of recombinant products/coagulation factors – recombinant products with extended half‑life.
Summary:
Current treatment of hemophilia is effective and safe. Patients´ quality of life has improved significantly in last two decades. With the era of the third generation recombinat coagulation factor concentrates, which are manufactured with no animal or human proteins, the risk of blood‑born infections was eliminated. The newest possibility of treatment are recombinant products with extended half‑life that is 1,5 times longer than standard recombinant products for the treatment of hemophilia A and 3–6 times for hemophilia B. Development of inhibitor – antibodies directed against FVIII/FIX products – remains, however, the most serious complication of the hemophilia treatment.
Úvod
Léčba hemofilie prošla za posledních šedesát let velkými změnami. V padesátých a šedesátých letech minulého století byly krvácivé epizody pacientů léčeny buď plnou krví, nebo čerstvě zmraženou plazmou. Vzhledem k nízkému obsahu koagulačních faktorů VIII a IX (FVIII a FIX) v těchto přípravcích pacienti s těžkou formou hemofilie často zemřeli již v dětském či v mladém dospělém věku převážně v důsledku krvácení při úrazech, zejména při poranění mozku a/nebo vnitřních orgánů či během operačního výkonu [1,2].
Zásadní změna v léčbě hemofilie nastala v roce 1964 poté, co Judith Pool objevila, že kryoprecipitát frakcionovaný z čerstvé plazmy obsahuje vysoké množství FVIII [1,2]. Celosvětově se počátek moderní léčby hemofilie datuje do sedmdesátých let dvacátého století, kdy začaly být vyráběny lyofilizované plazmatické deriváty FVIII a FIX [3]. Vedle účinnější léčby hemofilie došlo také k rozvoji domácí léčby bez nutnosti hospitalizace při každém krvácení. V České republice jsou plazmatické deriváty dostupné od devadesátých let.
Během sedmdesátých a osmdesátých let se mnoho pacientů s hemofilií nakazilo virem HIV či hepatitidy C. Příčinou byly kontaminované plazmatické deriváty získané ze směsné plazmy tisíců dárců [1,2]. Následně zavedené techniky virové inaktivace ve výrobě plazmatických derivátů a screening dárců výrazně zlepšily bezpečnost plazmatických derivátů, což potvrzuje i fakt, že za posledních dvacet let nedošlo k další nákaze [1,4].
Rozvoj DNA technologií v osmdesátých letech umožnil průmyslovou výrobu rekombinantních derivátů FVIII a FIX [5]. První rekombinantní FVIII byl pro léčbu dostupný v roce 1992 a FIX v roce 1997. V České republice jsou rekombinantní deriváty k dispozici od roku 2006. Rekombinantní deriváty první generace jsou vyráběny pomocí technologií rekombinantní DNA v ovariálních buňkách křečíka čínského (chinese hamster ovary cell, CHO) nebo v buňkách ledvin mláďat křečků (baby hamster kidney cell, BHK) a v tkáňových kulturách obsahují bovinní proteiny. Navíc jsou stabilizovány lidským albuminem. Druhá generace rekombinantního FVIII (rFVIII) je derivovaná z BHK a místo albuminu obsahuje sacharózu. Nicméně frakce lidských plazmatických proteinů používaná pro médium tkáňové kultury není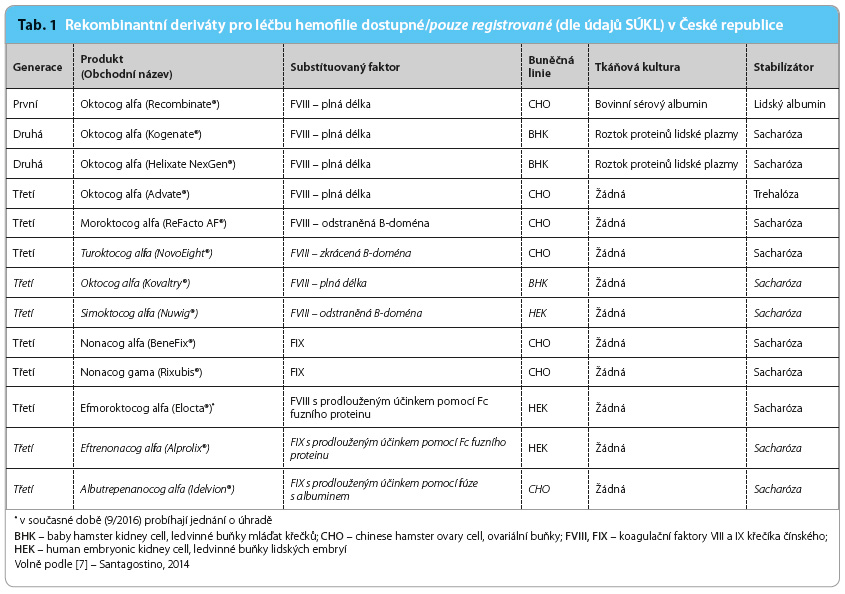 vysoce purifikovaná, a deriváty proto mohou obsahovat stopy lidských plazmatických bílkovin. Rekombinantní FVIII a FIX třetí generace neobsahují ani lidské, ani zvířecí plazmatické proteiny a jsou produkovány na CHO, BHK a na buňkách ledvin lidských embryí (human embryonic kidney cell, HEK) [6,7]. Nejnovějšími přípravky k léčbě hemofilie typu A i B jsou rekombinantní deriváty třetí generace s prodlouženým biologickým poločasem eliminace (t1/2). Podrobný přehled rekombinantních derivátů dostupných či registrovaných v České republice je uveden v tab. 1.
vysoce purifikovaná, a deriváty proto mohou obsahovat stopy lidských plazmatických bílkovin. Rekombinantní FVIII a FIX třetí generace neobsahují ani lidské, ani zvířecí plazmatické proteiny a jsou produkovány na CHO, BHK a na buňkách ledvin lidských embryí (human embryonic kidney cell, HEK) [6,7]. Nejnovějšími přípravky k léčbě hemofilie typu A i B jsou rekombinantní deriváty třetí generace s prodlouženým biologickým poločasem eliminace (t1/2). Podrobný přehled rekombinantních derivátů dostupných či registrovaných v České republice je uveden v tab. 1.
Rekombinantní deriváty třetí generace
Jak již bylo zmíněno v úvodu, deriváty třetí generace pro léčbu hemofilie se připravují bez přidání jakéhokoliv exogenního lidského či zvířecího proteinu během buněčné kultivace, purifikace nebo finální formulace. V České republice jsou nyní dostupné dva přípravky pro léčbu hemofilie A a dva pro léčbu hemofilie B. Přípravky pro léčbu hemofilie A se liší počtem aminokyselin, které rFVIII obsahuje. Na rozdíl od oktocogu alfa má moroktocog alfa odstraněnu tzv. B doménu (908 aminokyselinových zbytků), která není nutná pro vlastní proces krevního srážení, ale je důležitá pro intracelulární transport a pro sekreci FVIII, nicméně kompletní či částečná delece B domény se nezdá být spojena se signifikantním rozdílem v prokoagulačním účinku [8]. Počet i zastoupení aminokyselin v obou dostupných rFIX je stejný.
Stejně jako předchozí přípravky se deriváty třetí generace u pacientů s těžkou hemofilií používají nejen k profylaxi primární (podání derivátu po prvním kloubním či větším krvácení v měkkých tkáních před ukončeným druhým rokem života) a sekundární (profylaktické podávání derivátu započaté později než při primární profylaxi nebo po větším počtu krvácení), ale i k léčbě krvácení či při operaci, a to i u pacientů se střední či lehkou hemofilií. U dospělých pacientů je dávka při profylaktické léčbě hemofilie A 25–40 IU/kg 2–3× týdně a 1–2× týdně u pacientů s hemofilií B. U dětí dosud neléčených (previously untreated patients, PUPs) se preferuje zahájení léčby dávkou 25–50 IU/kg podávanou 1× týdně a následně dle počtu krvácení se dávkování postupně zvyšuje až ke standardnímu protokolu 3× týdně [9].
Dávka podaného derivátu při úrazu či při operačním výkonu závisí na tíži hemofilie, na závažnosti krvácení a na typu plánovaného výkonu. V těchto situacích se vychází z faktu, že 1 IU rFVIII na 1 kg tělesné hmotnosti zvýší aktivitu plazmatického FVIII o 2 % a 1 IU rFIX na 1 kg tělesné hmotnosti zvyšuje aktivitu plazmatického FIX přibližně o 1 % [9,10]. Při mírném a středně těžkém krvácení (epistaxe, krvácení do kloubů a svalů) je vhodné dosáhnout aktivity FVIII 40–60 % a FIX 30–50 %, při závažném krvácení (CNS, musculus iliopsoas či jakékoliv jiné masivní krvácení) je požadovaná aktivita FVIII 80–100 % a FIX 60–80 %. Doporučené hodnoty aktivity FVIII a FXI u jednotlivých typů krvácení či operačních výkonů jsou uvedeny v tab. 2.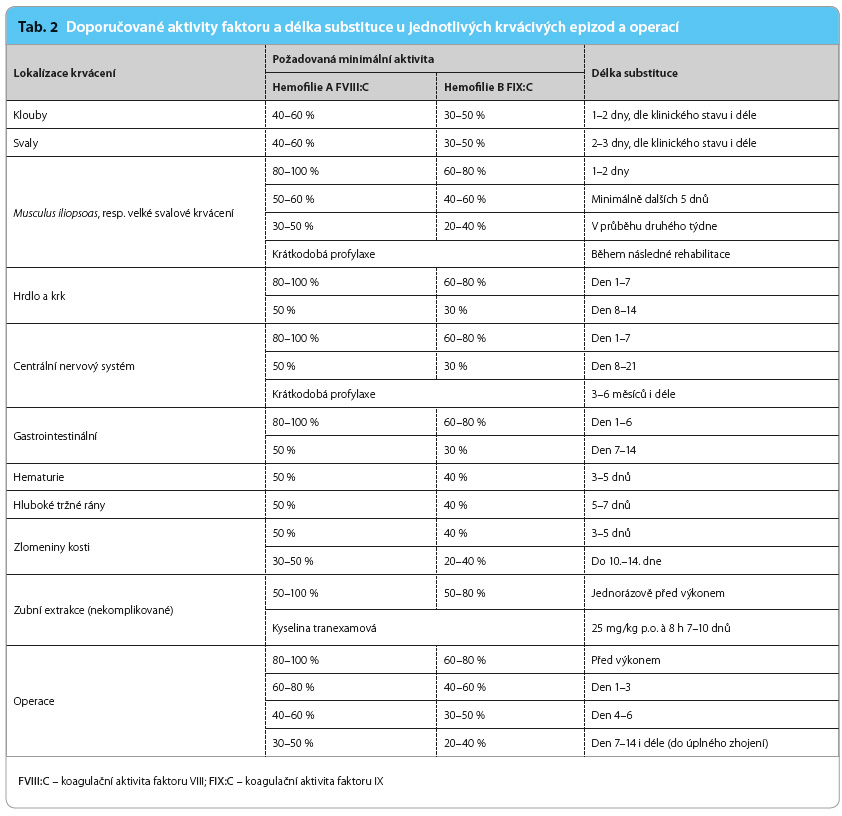
U dětí a zejména u novorozenců je vhodné oproti dospělým vzhledem k rozdílnému t1/2 navýšit substituci koncentráty FVIII/FIX o zhruba 10–25 %. Základním pravidlem při léčbě hemofilie v případě podezření na krvácení ohrožující život či končetinu je nejdříve substituovat faktor a teprve potom provádět vyšetření k potvrzení diagnózy (zejména při suspektním krvácení do CNS) [9].
Nejzávažnější komplikací léčby hemofilie je vznik inhibitoru (polyklonální protilátky IgG) proti podávanému faktoru, k čemuž dochází asi u 25–30 % pacientů s těžkou hemofilií A a u 4–5 % pacientů trpících hemofilií B [11,12]. K eradikaci inhibitoru je nutno zavést tzv. imunotoleranční léčbu. Pro tento typ léčby jsou dostupná různá léčebná schémata s rozpětím dávek od 50 IU/kg FVIII/FIX podávaných každý druhý den až po 200 IU/kg denně a jejich výběr pro konkrétního pacienta se řídí příslušnými doporučenými postupy [13]. V době výskytu inhibitoru jsou krvácivé epizody zajištěny přípravky, které obcházejí aktivitu FVIII a FIX, tj. aktivovaným rekombinantním faktorem VII (rFVIIa) nebo koncentrátem aktivovaného protrombinového komplexu (aPCC). Tyto přípravky se pro svůj krátký t1/2 k běžné profylaxi tak často nepoužívají [14].
V nedávné době byly publikovány výsledky studie Sippet [15], která uvádí, že výskyt inhibitoru je vyšší po podání rekombinantních koncentrátů FVIII ve srovnání s podáním plazmatických koncentrátů FVIII. Tento fakt je ale třeba dát do souvislosti mimo jiné právě s vyšší virovou bezpečností rFVIII. Diskuse o významu a interpretaci této zatím ojedinělé studie v současnosti probíhá.
Rekombinantní deriváty třetí generace s prodlouženým biologickým poločasem eliminace
Nejnovější možností léčby hemofilie jsou rekombinantní deriváty s prodlouženým biologickým poločasem eliminace, které jsou produkovány na buňkách HEK. U přípravků pro léčbu hemofilie typu A je t1/2 1,5–1,6× delší než u doposud dostupných derivátů, aplikace tedy mohou probíhat přibližně každých 3–5 dní. U derivátů určených k léčbě hemofilie typu B je t1/2 prodloužen 3–6× [12] a přípravek se podává v týdenních až dvoutýdenních intervalech. Technologie umožňující prodloužení t1/2 představují pegylace, kdy se k vlastnímu léčivu kovalentně naváží polymery polyethylenglykolu [16–18], a fúze, při které se koagulační protein sloučí s proteinem s mnohem delším t1/2 [19,20], konkrétně s Fc fragmentem lidského neonatálního imunoglobulinu G nebo s rekombinantním lidským albuminem [14].
Příčinou toho, proč je t1/2 rFVIII výrazně kratší než u rFIX, je závislost t1/2 FVIII navázaného z 95–98 % v krevním oběhu na clearance von Willebrandova faktoru [21,22], jehož t1/2 je 12–20 hodin [23,24].
V některých zemích jsou tyto přípravky k dispozici od roku 2014. V České republice zatím pokračují klinické studie fáze III a IV, je zaregistrován jeden přípravek obsahující derivát pro léčbu hemofilie A a dva pro léčbu hemofilie B. U přípravku pro léčbu hemofilie A v současné době již probíhají jednání o jeho úhradě.
Závěr
Možnosti léčby hemofilie jsou v dnešní době široké, terapie je bezpečná a účinná. Zavedení domácí a profylaktické léčby snižuje riziko vzniku krvácení a pacientům umožňuje plnohodnotný život. Limitem doposud dostupných derivátů pro léčbu hemofilie je jejich krátký t1/2 (8–12 hodin u FVIII a 18–24 hodin u FIX) a s tím související relativně častá nitrožilní aplikace. To může být zvláště u pediatrických pacientů závažným problémem podstatně snižujícím kvalitu života. U malých dětí, zejména u kojenců a batolat, kteří také hůře snášejí vlastní aplikace, je proto často nezbytné zavést na přechodnou dobu centrální žilní přístup, např. port.
Riziko onemocnění jakoukoliv infekcí přenosnou krví se díky rekombinantním derivátům třetí generace eradikovalo, avšak stále přetrvává riziko vzniku inhibitoru.
Seznam použité literatury
- [1] Franchini M, Mannuci PM. Past, present and future of hemophilia: a narrative review. Orphanet J Rare Dis 2012; 7: 24.
- [2] Mannuci PM. Back to the future: a recent history of hemophilia treatment. Hemophilia 2008; 14 (Suppl 3):
- [3] Webster WP, Roberts HR, Thelin GM, et al. Clinical use of new glycine‑precipitated antihemophilic fraction. Am J Med Sci 1965; 250: 643–651.
- [4] Tabor E. The epidemiology of virus transmission by plasma derivates: clinical studies verifying the lack of transmission of hepatitis b and C and HIV type 1. Transfusion 1999; 39: 1160–1168.
- [5] Morfini M, Coppola A, Franchini M, Di Minno G. Clinical use of factor VIII and factor IX concentrates. Blood Transfus 2013; 11 (Suppl 4): 55–63.
- [6] Franchini M, Lippi G. Recombinant FVIII concentrates. Semin Thromb Hemost 2010; 36: 493–497.
- [7] Santagostino E. A new recombinant factor VIII: from genetics to clinical use. Drug Des Devel Ther 2014; 8: 2507–2515.
- [8] Pipe SW. Functional roles of factor FVIII B domain. Hemophilia 2009; 15: 1187–1196.
- [9] Blatný J, Hrachovinová I, Hrdličková R, et al. Diagnostika a léčba hemofilie. Český národní hemofilický program 2012.
- [10] Srivastava A, Brewer AK, Mauser‑Bunschoten EP, et al. Treatment Guidelines Working Group on Behalf of The World Federation of Hemophilia. Guidelines of Management of Hemophilia. Hemophilia 2013; 19: 1–47.
- [11] Franchini M, Mannucci PM. Inhibitors of propagation of coagulation (factors VIIIm IX and XI): a review of current therapeutic practice. Br J Clin Pharmacol 2011; 72: 553–562.
- [12] Peyvandi F, Garagiola I, Young G. The past and future of hemophilia: diagnosis, treatments
- [13] Hay CR, DiMichele DM. International Immune Tolerance Study. The principal results of The International Immune Tolerance Study: a randomized dose comparison. Blood 2012; 119: 1335–1344.
- [14] Shetty S, Ghosh K. Novel therapeutic approaches for haemophilia. Haemophilia 2015; 21: 152–161.
- [15] Peyvandi F, Mannucci PM, Garagiola I, et al. A Randomized Trial of Factor VIII and Neutralizing Antibodies in Hemophilia A. N Engl J Med 2016; 374: 21.
- [16] Tiede A, Brand B, Fischer R, et al. Enhancing the pharmacokinetic properties of recombinant factor VIII: first‑in‑man trial of glycoPEGylated recombinant factor VIII in patients with hemophilia A. J Thromb Haemost 2013; 11: 670–678.
- [17] Coyle TE, Reding MT, Lin JC, et al. Phase I study of Bay 94‑9027, a PEGylated B‑domain‑deleted recombinant factor VIII with an extended half‑life, in subject with hemophilia A. J Thromb Haemost 2014; 12: 488–496.
- [18] Collins PW, Young G, Knobe K, et al. Recombinant long‑acting glykoPEGylated factor IX in hemophilia B: a multinational randomized phase 3 trial. Blood 2014; 124: 3880–3886.
- [19] Mahlangu J, Powel JS, Ragni MV, et al. Phase 3 study of recombinant factor VIII Fc fusion protein in severe hemophilia A. Blood 2014; 133: 317–325.
- [20] Martinowitz U, Lissitchow T, Tubetsky A, et al. Result of a phase I/II open‑label, safety and efficacy trial of coagulation factor IX (recombinant), albumin fusion protein in haemophilia B patients. Haemophilia 2015; 21: 784–790.
- [21] Tang L, Leong L, Sim D, et al. von Willebrand factor contributes to longer half‑life of PEGylated factor VIII in vivo. Haemophilia 2013; 19: 539–545.
- [22] Denis CV, Christophe OD, Oortwijn BD, Lenting PJ. Clearance of von Willebrand factor. Thromb Haemost 2008; 99: 271–278.
- [23] Robertson J, Lillicrap D, James PD. Von Willebrand Disease. Pediatr Clin N Am 2008; 55: 377–392.
- [24] Sadler JE, Budde U, Eikenboom JC, et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the subcommittee on von Willebrand factor. J Thromb Haemost 2006; 4: 2103–2114.
- [25] Salaj P, Smejkal P, Komrska V, et al. Standardy péče o nemocné s hemofilií. Český národní hemofilický program 2012.