Anticholinergní látky v léčbě hyperaktivního močového měchýře
Hyperaktivní močový měchýř je symptom, který se vyskytuje u velkého počtu pacientů. U starších lidí představuje jedno z nejčastějších onemocnění. Prevalence onemocnění narůstá se zvyšujícím se věkem (vyskytuje se přibližně u 17 % lidí starších 18 let, ve věku nad 75 let trpí hyperaktivním močovým měchýřem již 31 % žen a 42 % mužů). Hlavní roli v léčbě hyperaktivního močového měchýře má anticholinergní léčba. Blokáda cholinergních receptorů snižuje výskyt příznaků, jako je urgence, časté močení, inkontinence a nykturie. Cholinergní receptory jsou tvořeny 5 subtypy receptorů (M1–M5). Močový měchýř obsahuje jak receptory M2, tak receptory M3. Receptor M3 hraje hlavní roli při kontrakci močového měchýře a při vzniku některých patologických stavů. Nevýhodou anticholinergní léčby je výskyt nežádoucích příznaků, mezi které patří suchost v ústech, mlhavé vidění, zácpa a jiné. Vlastnosti anticholinergních látek záleží na jejich receptorové selektivitě, která ovlivňuje jejich účinnost a výskyt nežádoucích účinků. Bezpečnost anticholinergních látek závisí také na jejich farmakokinetice, metabolismu, vazbě na bílkoviny a schopnosti pronikat hematoencefalickou bariérou. Nově zaváděné anticholinergní látky vykazují vyšší selektivitu a vysokou afinitu k podtypům muskarinových receptorů uložených v močovém měchýři. V mnoha klinických randomizovaných studiích bylo prokázáno, že selektivní aktivace receptorů je významnou podmínkou snášenlivosti anticholinergik.
Úvod
Hyperaktivní močový měchýř (OAB – overactive bladder) je podle Mezinárodní společnosti pro kontinenci (ICS – International Continence Society) definován jako soubor příznaků dysfunkce dolních močových cest. K příznakům typickým pro OAB patří urgence, která může být spojena i s inkontinencí. Urgence je definována jako náhlý nepřekonatelný pocit nucení na močení. Dalšími příznaky jsou zvýšená frekvence močení a nykturie. Hyperaktivní močový měchýř má výrazný negativní dopad na kvalitu života pacientek. I v případech, že je lékař informován o příznacích OAB, pacientka často nedostane odpovídající léčbu. Mezinárodní společnost pro kontinenci určila čtyři základní příznaky hyperaktivního močového měchýře:
– ‑urgence (nucení na močení) – náhlý a nutkavý pocit nutnosti vymočit se, který je obtížné oddálit,
– ‑inkontinence – každý nechtěný únik moče,
– ‑časté močení (polakisurie) – příliš časté nucení na močení (8 a více mikcí za den),
– ‑nykturie – probuzení jednou nebo vícekrát za noc kvůli nutnosti močení.
Hlavním příznakem hyperaktivního močového měchýře je nadměrné nucení (urgence). Urgence se na rozdíl od fyziologického nucení dostavuje náhle a nutí pacientku k rychlému vymočení se. Zkracuje se doba od prvního pocitu na močení k eventuálnímu spontánnímu úniku moče, zvyšuje se frekvence a snižují se objemy mikce. Urgence může být doprovázena i únikem moče, přesto dvě třetiny pacientů s OAB inkontinenci nemají.
Fyziologie funkce močového měchýře
Několik základních fyziologických mechanismů se účastní na regulaci funkce močového měchýře. Plnění močového měchýře a močení je primárně kontrolováno stimulací nerovových parasympatických a sympatických zakončení ve stěně a hrdle močového měchýře [1].
Při plnění močového měchýře (relaxace stimulovaná sympatikem) je detruzor relaxovaný a zároveň je tonizované (kontrahované) hrdlo močového měchýře. Během mikce (kontrakce vyvolané parasympatikem) je detruzor kontrahovaný a hrdlo močového měchýře relaxované. Aktivace b-adrenergních receptorů noradrenalinem je odpovědná za relaxaci vyvolanou sympatikem, která umožní plnění močového měchýře. Aktivace muskarinových receptorů acetylcholinem vyvolá kontrakci, která vyústí v mikci.
Urgence může být způsobena hyperaktivitou detruzoru, která se projevuje přítomností typických nechtěných kontrakcí při plnící fází během urodynamického vyšetření. Kontrakce mohou vzniknout spontánně nebo mohou být vyprovokovány.
Tento stav byl popisován termínem nestabilita detruzoru. Podle nové klasifikace ICS byl tento termín nahrazen termínem idiopatická hyperaktivita detruzoru.
Příčinou je nedostatečné nebo porušené centrální tlumení mikčního centra. Při normální senzorické aferentní signalizaci z močového traktu zde vznikají neinhibované kontrakce detruzoru.
Kontrakce detruzoru se projevují na cystometrické křivce jako vlny, které jsou buď jednotlivé, mnohočetné nebo v salvách. Detruzorové kontrakce zvyšují intravezikální tlak nejméně o 15 cm H2O, často jsou i vyšších hodnot. Tato dysfunkce detruzoru může být provázena zmenšenou kapacitou měchýře.
Idiopatická hyperaktivita detruzoru je druhou nejčastější příčinou inkontinence moče a je prokázána až u 40 % pacientek indikovaných k urodynamickému vyšetření. Předpokládá se, že tato porucha postihuje až 10 % populace. Incidence idiopatické hyperaktivity detruzoru se zvyšuje s věkem a ve stáří je nejčastější příčinou inkontinence. Neinhibované kontrakce detruzoru jsou příčinou polakisurie, nykturie, urgence (imperativního nucení na močení).
Dosud není plně objasněna patofyziologie tohoto onemocnění, a proto jeho léčba často selhává. U malého procenta žen se prokáže neurologická příčina obtíží. Na doporučení ICS jsou případy podmíněné neurologickými příčinami zahrnovány do skupiny neurogenní hyperaktivity detruzoru (dříve hyperreflexního měchýře) [2, 3]. Neurofyziologickou podstatou neurogenní hyperaktivity (hyperreflexie) jsou suprasakrální neurologické léze, které způsobí nedostatečnou kontrolu mikčního reflexu. Již slabé podněty z proprioceptivních receptorů vyvolávají detruzorovou kontrakci. Hyperaktivita detruzoru může být i následkem špatného zvládnutí kontroly močového měchýře v dětství. Behaviorální techniky jsou často úspěšné při léčbě poruchy. Také byla prokázána souvislost mezi enuresis nocturna v dětství a rozvojem idiopatické hyperaktivity detruzoru v dospělosti.
Velmi často je diskutována i psychická dispozice ke vzniku onemocnění. Výsledky různých studií jsou rozdílné a rozporuplné. Walters na základě psychometrického vyšetření neprokázal rozdíly mezi ženami se stresovou inkontinencí a hyperaktivitou detruzoru [4]. Při srovnání s kontrolní skupinou měly ženy s hyperaktivitou detruzoru vyšší skóre pro deprese, hypochondrii a hysterii. V jiné studii založené na psychiatrickém vyšetření se neprokázal rozdíl v psychiatrické morbiditě mezi ženami se stresovou inkontinencí a nestabilitou detruzoru [5]. Zajímavostí této studie bylo, že ženy s negativním urodynamickým nálezem měly nejvyšší skóre pro strach a úzkost, které bylo srovnatelné s nálezem u psychiatrických pacientů.
Možnou příčinou vzniku abnormálních kontrakcí detruzoru je primární patologie v oblasti uretry. Abnormální funkce může být způsobena obstrukcí nebo hyperaktivitou uretry nebo se uretra nemůže otevřít v důsledku anatomické abnormality.
Další možnou příčinou je detruzor-sfinkterová dyssynergie. Při tomto onemocnění se prokazují zároveň kontrakce detruzoru s mimovolním stahem uretrálního nebo periuretrálního příčně pruhovaného svalu.
Podle integrální teorie je další příčinou vzniku idiopatické hyperaktivity detruzoru insuficience hrdla močového měchýře. Moč proniká do proximální uretry, což následně vyvolá neinhibované kontrakce detruzoru.
Další příčinou urgentní inkontinence jsou etiologicky idiopatické formy s normálním neurologickým nálezem, které mohou mít původ v samotném měchýři, např. chronická recidivující cystitida, atrofie sliznice močového měchýře, „svráštělý" měchýř po ozařovaní, tumory měchýře, cizí tělesa v močovém měchýři, divertikl, velká cystokéla aj.
Senzorickou urgenci diagnostikujeme na základě urodynamického vyšetření. Postižené ženy si stěžují na zvýšenou frekvenci mikce, urgenci nebo urgentní inkontinenci a při urodynamickém vyšetření není přítomna hyperaktivita detruzoru. Příznaky senzorické urgentní inkontinence jsou stejné jako u motorické urgentní inkontinence, pouze únik moče bývá méně častý.
Etiologie senzorické urgence není ještě plně objasněna. U senzorické urgentní inkontinence je mikční reflex vyvolán zesílenými aferentními impulzy z receptorů registrujících roztažení stěny močového měchýře při normální centrální motorické inhibici. Na vzniku se podílejí i psychologické faktory. Při urodynamickém vyšetření mohou být přítomny příznaky nízké compliance (vzestup tlaku v močovém měchýři je vyšší než 2,6 cm H20 na 100 ml náplně) nebo příznaky uretrální instability (kolísání uretrálního tlaku vyšší než 15 cm H2O v místě maximálního uzavíracího tlaku v uretře). Podle některých autorů lze použít následující diagnostická kritéria: první pocit urgence při plnění močového měchýře je při náplni nižší než 100 ml a kapacita močového měchýře je nižší než 400 ml. Vzhledem k vágním diagnostickým kritériím neznáme přesnou incidenci senzorické urgence (předpokládá se přibližně 10% incidence).
Senzorická urgentní inkontinence může být sekundární – vyskytuje se u zánětlivé nebo nádorové infiltrace stěny močového měchýře, u urolitiázy aj. Při zánětech močového měchýře jsou stimulovány proprioceptivní receptory, takže mikční reflex je vyvolán i minimálním současným podnětem i při malém objemu močového měchýře. Na rozvoji inkontinence u cystitis se může podílet i vliv endotoxinu E. coli, který má a-blokující efekt na oblast hrdla močového měchýře [6].
Léčba hyperaktivního močového měchýře
Léčba hyperaktivního močového měchýře je většinou konzervativní. Pokud známe příčinu vzniku OAB, snažíme se ji odstranit. Ve většině případů však volíme léčbu, snažíme se obnovit centrální kontrolu mikce nebo modifikovat inervaci. Léčebné postupy můžeme rozdělit do čtyř základních skupin:
– ‑ovlivnění chování močového měchýře
– ‑elektrická stimulace
– ‑chirurgická léčba
– ‑medikamentózní léčba.
Při ovlivnění funkce močového měchýře se snažíme o znovuzískání kortikální (CNS) kontroly močového měchýře. K získání kontroly můžeme využít trénink pravidelného močení, psychoterapii, hypnózu, biofeedback a akupunkturu.
Elektrická stimulace je založena na stimulaci aferentních vláken n. pudendus, což vyvolá i podráždění eferentních vláken a zvýší se kontraktilita příčně pruhovaného uretrálního svalu a svaloviny pánevního dna. Současně se ovlivňuje i vegetativní systém, inhibuje se účinek parasympatiku a stimulují se b-sympatické receptory ve stěně močového měchýře.
Chirurgická léčba se běžně nepoužívá. V praxi se nejčastěji provádí distenze močového měchýře.
Medikamentózní léčba
V terapii hyperaktivního močového měchýře je nejúspěšnější medikamentózní léčba. Současné znalosti o neurofyziologii a vegetativní inervaci močového měchýře umožňují účinnější výběr léku. Léčebný efekt farmakologické léčby se pohybuje mezi 60–80 %. Základní skupinou pro léčbu OAB jsou anticholinergní látky, kterým bude věnována i většina následujícího textu. Účinnost těchto preparátů je limitována nedostatečným selektivním zaměřením na močový měchýř, což je příčinou nežádoucích vedlejších účinků, které mohou vést až k přerušení léčby.
V praxi se při léčbě OAB také používají tricyklická antidepresiva (imipramin), lokální aplikace estrogenů a intravezikální aplikace botulotoxinu (pouze u vybraných pacientek po selhání ostatní konzervativní léčby).
Ostatní skupiny léčiv se v praxi zatím neuplatňují (blokátory vápníkových kanálů, inhibitory syntézy prostaglandinů, antiparkinsonika, látky ovlivňující draslíkový kanál, b2-sympatomimetika, a-sympatolytika, a-sympatomimetika) [7].
Anticholinergní látky
Chemické vlastnosti anticholinergních látek
Anticholinergní látky, které se používají při léčbě hyperaktivního močového měchýře, patří k amoniovým derivátům, kde je jeden nebo více atomů vodíku nahrazen alkylovou nebo arylovou skupinou. Amoniové sloučeniny pak dělíme na primární, sekundární, terciární a kvarterní podle počtu substituovaných vodíků. Kvarterní amoniové sloučeniny (trospium chlorid) mají hydrofilní vlastnosti, špatně se rozpouštějí v tucích, a proto i špatně pronikají lipidovou buněčnou membránou. Kvarterní amoniové sloučeniny se špatně resorbují z GIT, mají nízkou biologickou dostupnost a nepronikají hematoencefalickou bariérou.
Oxybutynin, tolterodin, darifenacin, solifenacin a propiverin jsou terciární amoniové sloučeniny. Hlavní rozdíl mezi terciárními a kvarterními sloučeninami představují hydrofilní vlastnosti kvarterních sloučenin. Terciární sloučeniny jsou lipofilní, mají vyšší biologickou dostupnost, ale také snadněji pronikají hematoencefalickou bariérou než sloučeniny kvarterní [8–11].
Muskarinové receptory
Četné studie prokázaly přítomnost specifických muskarinových receptorů (M1-M5) v různých lokalizacích: M1 – v nervové soustavě; M2 – v srdci a v detruzoru; M3 – v detruzoru, ve slinných žlázách a v jiných žlázách se sekretorickou funkcí a v GIT; M4 – v mozkové kůře a plicích; M5 – dosud není známo. Přítomnost všech těchto receptorů byla prokázána i v CNS [1, 12–14].
Receptory M2 a M3 jsou přítomny v močovém měchýři. Receptory M3 se přímo podílejí na kontrakci hladkého svalu. Receptory M2 působí synergicky s receptory M3 a umožní efektivnější vypuzení moče.
Receptory M3 vyvolávají kontrakci detruzoru převážně stimulací hydrolýzy fosfoinositolu, která vede k uvolnění intracelulárního kalcia. Studie na zvířecím modelu prokázaly, že receptory M2 se na kontrakci podílejí inhibicí cAMP, jež se podílí na relaxaci svalu. Na vyprázdnění močového měchýře působí dvojí mechanismus – kontrakce detruzoru vyvolaná receptory M3 a relaxace detruzoru zablokovaná receptory M2. Tento mechanismus fyziologicky umožní kompletní vyprázdnění močového měchýře.
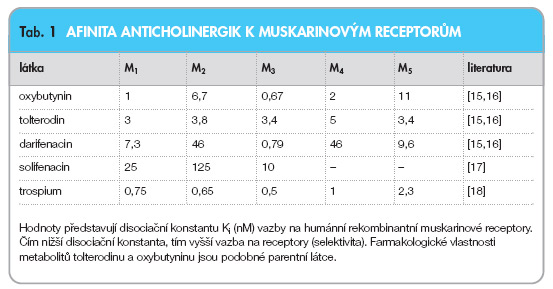
V tab. 1 je uvedena selektivita anticholinergních látek k muskarinovým receptorům užívaných v léčbě OAB. Vazebná afinita však nemusí zcela korelovat s aktivitou in vivo.
Neselektivní anticholinergní látky
Antimuskarinové látky dosahují své účinnosti vazbou na muskarinové receptory, blokují aktivitu acetylcholinu a snižují kontrakční aktivitu detruzoru. Nicméně tyto látky ovlivňují muskarinové receptory i v jiných tkáních (slinná žláza, gastrointestinální trakt) [19, 20]. V preklinických studiích bylo prokázáno, že tolterodin a oxybutynin se stejně váží na muskarinové receptory v močovém měchýři, ale oxybutynin má 8krát vyšší afinitu k receptorům ve slinné žláze než tolterodin. Ve studiích in vivo u koček bylo také prokázáno, že tolterodin má vyšší inhibiční efekt na kontrakce detruzoru než na ovlivnění salivace [15, 16].
Trospium chlorid má malou selektivitu, ale vysokou celkovou afinitu ke všem podtypům muskarinových receptorů [21]. Solifenacin se váže na receptory M1, M2 a M3, ale prokazuje vyšší afinitu k muskarinovým receptorům v močovém měchýři než ve slinné žláze (podobně jako tolterodin) [22]. Studie provedené u zvířat také předpokládají, že trospium chlorid vykazuje vyšší stupeň selektivity k močovému měchýři in vitro a in vivo než tolterodin a oxybutynin [23, 24].
Selektivní anticholinergní látky
Darifenacin je první M3-selektivní antimuskarinová látka pro léčbu hyperaktivního močového měchýře [25]. Laboratorní studie dokazují, že má vyšší selektivitu pro močový měchýř než pro slinnou žlázu [26].
Farmakokinetické vlastnosti anticholinergních látek
Absorpce
Biologická dostupnost odpovídá frakci nezměněného léku, který se po aplikaci dostane do systémového oběhu. Neúplná absorpce je jedním z hlavních faktorů, který ovlivňuje biologickou dostupnost léčiva. K ostatním faktorům, které ovlivňují biologickou dostupnost léčiva, patří ovlivnění vstřebávání léku v závislosti na příjmu potravy.
Anticholinergika se čtyřmocným dusíkem v molekule mají obecně špatnou biologickou dostupnost. Vzhledem k jejich hydrofilitě dostupnost kolísá v závislosti na typu použitého preparátu, pro trospium se udává biologická dostupnost po perorálním podání kolem 10 %. Na druhé straně má tolterodin biologickou dostupnost vyšší než 74 %. Sníženou biologickou dostupnost můžeme obejít zvýšeným dávkováním léku a jeho aplikací nalačno, bez signifikantního zvýšení počtu nežádoucích účinků.
Metabolismus
Většina léčiv se odbourává v játrech prostřednictvím cytochromu P-450. Léčiva, která se odbourávají stejným izoenzymem, se mohou kompetitivně vázat na stejné vazebné místo. Léčivo, které má vyšší afinitu k vazebnému receptoru, bude odbouráno mnohem rychleji než léčivo s nižší afinitou. Tímto mechanismem se může porušit jejich absorpce, distribuce, eliminace, což může vést ke zvýšené nebo snížené koncentraci léku v místě předpokládaného působení. Interakce mohou být závažné u pacientů, kteří užívají více léčiv najednou.
Oxybutynin a tolterodin jsou metabolizovány systémem enzymů cytochromu P-450, oxybutynin izoenzymem CYP3A4 a tolterodin CYP2D6. V naší populaci lze předpokládat deficienci CYP2D6 u 7–10 % lidí, což může být příčinou rozdílného účinku u různých lidí.
Na druhé straně trospium chlorid se prakticky nemetabolizuje systémem enzymů cytochromu P-450. Tím se výrazně snižuje riziko lékových interakcí s ostatními léky. Další důležitou vlastností trospium chloridu je, že se látka v 80 % nezměněná vylučuje do moče. Tolterodin a oxybutynin se naopak v nezměněné podobě vylučují do moče v méně než 5 %. Akumulace trospia v močovém měchýři může ještě zvýšit distribuci aktivní látky do močového měchýře, a zvýšit tak efekt léčby. Tímto způsobem se může vysvětlit snížené množství celkových nežádoucích účinků a zvýšená snášenlivost ve srovnání s ostatními léky. Solifenacin se významně metabolizuje v játrech, především systémem cytochromu CYP3A4, 70 % solifenacinu se vylučuje do moči, 23 % do stolice. Darifenacin je významně metabolizován cytochromy CYP3A4 a CYP2D6 v játrech a CYP3A4 ve stěně střevní. Látka se vylučuje ze 60 % močí a ze 40 % do stolice.
Řada studií prokázala po intravezikální aplikaci anticholinergních látek sníženou frekvenci nežádoucích účinků [27–29].
Biologický poločas
Biologický poločas je doba potřebná k eliminaci 50 % léčiva z organismu. Množství dostupného léčiva částečně závisí i na jeho biologickém poločase a na dávkovacím schématu. Dlouhý biologický poločas solifenacinu (45–68 hodin) umožňuje podávání 1krát denně; ostatní látky mají poločas výrazně kratší. Proto buď musí být podávány vícekrát denně (trospium chlorid, oxybutynin, tolterodin), nebo jsou vyráběny v tabletách s prodlouženým uvolňováním (darifenacin, tolterodin) nebo v náplastech (oxybutynin).
Bezpečnost
Anticholinergní látky mají po perorální aplikaci různé systémové vedlejší účinky. Nejčastějším vedlejším účinkem anticholinergní léčby OAB je suchost v ústech, poruchy akomodace a ovlivnění pasáže v oblasti GIT.
Nežádoucí účinky na CNS jsou obzvlášť závažné u starých lidí. Zmatenost a mentální poruchy mohou být příčinou nemožnosti aplikace anticholinergních látek. Anticholinergní látky jsou také kontraindikovány u lidí s glaukomem s ostrým úhlem, s tachykardií, s ulcerativní kolitis, myasthenia gravis, achalasií, obstrukcí GIT.
Kvarterní amoniové sloučeniny mají odlišnou frekvenci nežádoucích účinků. Při léčbě trospiem byla pozorována oproti léčbě oxybutyninem nižší frekvence výskytu suchosti v ústech. Po aplikaci trospia nebyly prokázány změny na EKG. Podobné vlastnosti má i tolterodin.
Nižší frekvenci nežádoucích účinků mají i lékové formy s postupným uvolňovaním (tolterodin, darifenacin, oxybutynin). Dalším významným faktorem, který ovlivňuje výskyt nežádoucích účinků, je tkáňová selektivita (novější látky – solifenacin a darifenacin).
Přehled vlastností jednotlivých látek
Oxybutynin s rychlým uvolňováním
V již dříve provedených studiích byla prokázána klinická účinnost perorálně podávaného oxybutyninu IR při redukci příznaků hyperaktivního močového měchýře, jako je urgence a urgentní inkontinence [30, 31].
Při srovnání s podáváním placeba léčba oxybutyninem výrazně snižuje celkový počet mikcí (4,5krát v. 5,7krát za den, p < 0,01) a urgence (9 v. 13 epizod, p < 0,01). Pacienti léčení oxybutyninem měli také výrazně snížený počet epizod urgentní inkontinence (pokles z 21 na 9, p < 0,001). Oxybutynin byl při redukci epizod urgentní inkontinence výrazně účinnější než placebo. Výrazné zlepšení při léčbě oxybutyninem oproti placebu bylo prokázáno i při objektivním měření, zmírnění naléhavosti prvního nucení na močení (p < 0,025), silného nucení na močení (p < 0,01) a detruzorového tlaku, který byl provázen silným nucením na močení (p < 0,02). Účinnost léčby oxybutyninem byla však doprovázena vysokou incidencí anticholinergních nežádoucích účinků. Nejčastějším nežádoucím účinkem byla suchost v ústech (73 % při léčbě oxybutyninem a 27 % při podávání placeba), poruchy zraku (33 % v. 17 %) a nauzea (33 % v. 13 %) [30].
V další dvojitě slepé studii byla srovnávána účinnost oxybutyninu (4krát 5 mg denně) s placebem. U pacientů léčených oxybutyninem bylo zaznamenáno významné zlepšení ve sledovaných parametrech oproti skupině pacientů, kteří dostávali placebo. I urodynamické vyšetření prokázalo statisticky významné zlepšení. U 31 z 37 pacientů, kteří užívali oxybutynin, se projevily nežádoucí účinky. Suchost v ústech se vyskytla u 94 % pacientů, u nichž se projevily vedlejší účinky léčby, a pouze u 32 % pacientů, kteří užívali placebo. Zácpa se vyskytla u 42 % léčených oxybutyninem a u 19 % pacientů užívajících placebo, suchá kůže se objevila u 32 % pacientů, kterým byl podáván oxybutynin, a pouze u 3 % pacientů dostávajících placebo, poruchy vizu u 23 % pacientů léčených oxybutyninem, a pouze u 3 % dostávajících placebo, nauzea u 23 % pacientů s léčbou oxybutyninem oproti žádnému pacientovi užívajícímu placebo. Při kontrole po 6 měsících užívalo oxybutynin už jen 7 ze 37 pacientů, a to pouze v redukované dávce [31].
Farmakologické vlastnosti
Oxybutynin je syntetické parasympatolytikum působící jako antagonista acetylcholinu na muskarinových receptorech, má slabý histaminový a lokálně anestetický účinek, působí spasmolyticky na hladkou svalovinu močového měchýře, neovlivňuje neuromuskulární ploténku ani vegetativní ganglia. Účinek léku nastupuje za 30–60 minut po podání a přetrvává 6–10 hodin. Maximální sérové hladiny je dosaženo během 30–60 minut po podání. V závislosti na trávení se oxybutynin resorbuje ze zažívacího traktu v tenkém střevě. Oxybutynin podléhá po vstřebání ve střevě významnému first-pass metabolismu, který vyústí ve vysokou koncentraci aktivních metabolitů. To vysvětluje, že cirkulující koncentrace N-desethyl-oxybutyninu (N-DEO) je přibližně 4–10krát vyšší než hladina původní látky. Oxybutynin i N-DEO se vysoce váží na plazmatické bílkoviny. N-DEO má signifikantně vyšší vazebnou afinitu ke slinné žláze než k močovému měchýři, což je příčinou vysoké incidence suchosti v ústech. Plazmatický eliminační poločas je dvoufázový, první fáze trvá 40 minut, druhá kolem 2–3 hodin. Eliminační poločas může být prodloužen u starších pacientů, zejména u nemocných se špatným celkovým zdravotním stavem. Oxybutynin a jeho metabolity jsou vylučovány do stolice a primárně do moče. Nebyla prokázána kumulace látky v organismu ani po opakovaném podávání. Podaná dávka je úplně vyloučena za méně než 8 hodin [32–35].
Oxybutynin s postupným uvolňováním
Bohužel, tato léková forma není v ČR k dispozici, proto následující popis bude zkrácený.
Studie, ve kterých byla vyhodnocována účinnost oxybutyninu ER, prokázaly účinnost v redukci příznaků hyperaktivního močového měchýře. Při srovnání s oxybutyninem IR došlo k významnému snížení výskytu nežádoucích účinků, hlavně suchosti v ústech [36–38].
V jiné prospektivní randomizované dvojitě slepé studii byla porovnána účinnost oxybutyninu ER s tolterodinem IR [39]. Pacienti léčení oxybutyninem měli při léčbě vyšší redukci urgentní inkontinence, snížený počet mikcí oproti pacientům léčeným tolterodinem. Snášenlivost obou režimů však byla identická. Po aplikaci lékové formy oxybutyninu s prodlouženým uvolňováním je oproti aplikaci formy s rychlým uvolňováním nižší maximální plazmatická koncentrace oxybutyninu a prodlužuje se doba k dosažení maximální plazmatické koncentrace. Snižuje se také plazmatická koncentrace primárního metabolitu N-DEO, prodlužuje se i doba eliminace, nejspíše díky zpomalené absorpci léku. Poměr oxybutyninu k jeho metabolitu N-DEO je vyšší u oxybutyninu s prodlouženým uvolňováním (0,40) než u preparátu s okamžitým uvolňováním (0,18) [36]. Sérové koncentrace N-DEO jsou nižší po perorální aplikaci oxybutyninu ER než po aplikaci IR formy. Tento rozdíl je způsoben absorpcí oxybutyninu, ke které dochází ve větší části GIT, částečně i v tlustém střevě, čímž dochází ke snížení first-pass efektu (oxidace v játrech).
Aplikace oxybutyninu nepodléhající first-pass efektu
Transdermální aplikace
Oxybutynin pro transdermální aplikaci opět není k dispozici v ČR. Po nalepení náplasti oxybutynin prostupuje do krevního řečiště a vyhýbá se tak first-pass metabolismu v GIT a v játrech, díky tomu se snižuje i koncentrace jeho metabolitu N-DEO a výrazně se mění poměr oxybutyninu k tomuto metabolitu oproti perorální aplikaci [40–42]. Změna poměru výrazně koreluje i se snížením výskytu nežádoucích účinků, které jsou vyvolány N-DEO.
Účinnost transdermální aplikace byla porovnána s perorální aplikací IR formy [43]. Nebyl prokázán rozdíl v účinnosti, ale po transdermální aplikaci byl zaznamenán signifikantně nižší výskyt nežádoucích účinků.
V jiné randomizované studii byla porovnávána efektivita léčby oxybutyninem v dávkách 1,3 mg/den, 2,6 mg/den a 3,9 mg/den oproti placebu [44]. Po aplikaci 3,9 mg oxybutyninu denně byl stejný výskyt nežádoucích účinků při léčbě transdermálním oxybutyninem jako při podávání placeba, suchost v ústech byla zaznamenána u 9,3 % pacientů ve skupině léčené oxybutyninem TDS a u 8,3 % pacientů ve skupině placeba, podobně závratě byly zaznamenány u 4 % nemocných ve skupině léčené oxybutyninem oproti 3,8 % u těch, kteří užívali placebo.
Nejčastějším vedlejším účinkem vyskytujícím se při transdermální aplikaci oxybutyninu byla lokální reakce v místě aplikace, nejčastěji s malou až střední intenzitou obtíží. Zarudnutí se objevilo u 5,6 % pacientů, kteří byli léčeni dávkou 3,6 mg oxybutyninu denně, u pacientů dostávajících placebo se zarudnutí vyskytlo ve 2,3 %. Pruritus se vyskytoval při léčbě oxybutyninem u 16,8 % nemocných, při podávání placeba u 6,1 % pacientů.
V další studii byl porovnán efekt transdermální aplikace oxybutyninu s tolterodinem ER a s placebem [45]. Při léčbě transdermálním oxybutyninem byly častější lokální reakce v místě aplikace, při léčbě tolterodinem byly častější celkové nežádoucí reakce. Nejčastěji se opět jednalo o pruritus ve 14 % (placebo 4,3 %), o zarudnutí v 8,3 % (placebo 1,7 %). Suchost v ústech se vyskytla u 4,1 % z celkového počtu pacientů léčených oxybutyninem, u 7,3 % z celkového počtu pacientů léčených tolterodinem a pouze u 1,7 % pacientů z těch, kteří dostávali placebo. Tato studie prokázala, že transdermální aplikace oxybutyninu má srovnatelnou účinnost s perorální aplikací, ale je spojena s nízkou incidencí celkových nežádoucích účinků provázejících anticholinergní léčbu.
Transdermální aplikace je vhodná pro pacienty, kteří nesnášejí perorální léčbu anticholinergními preparáty, užívají jinou perorální medikaci nebo nechtějí denně užívat medikaci.
Rektální aplikace oxybutyninu
Intrarektální aplikace je efektivní při léčbě příznaků OAB. Bohužel nejsou k dispozici komerčně připravené čípky. Účinnost byla prokázána v několika klinických studiích u pacientů nesnášejících perorální aplikaci [46]. Po intrarektální aplikaci oxybutynin neprochází portálním systémem, a proto je snížena produkce metabolitu N-DEO, což je příčinou nižšího výskytu nežádoucích účinků [47].
Intravezikální aplikace oxybutyninu
Intravezikální aplikace oxybutyninu (přímá aplikace roztoku či léku do močového měchýře) byla testována jako jedna z dalších z možností, jak se vyhnout first-pass efektu; ten při podání per os způsobuje zvýšený výskyt nežádoucích účinků vyvolaných metabolity oxybutyninu. Do močového měchýře byl aplikován roztok destilované sterilní vody, v němž byly rozpuštěny rozdrcené tablety 5 mg oxybutyninu. Roztok byl do močového měchýře aplikován katétrem. Tato metoda léčby se používá u pacientů s neurogenním močovým měchýřem, kteří vyžadují intermitentní katetrizaci močového měchýře.
Po intravezikální léčbě nebyly popsány nežádoucí účinky, významně se snížil výskyt inkontinence [48, 49]. Nevýhodou je nutnost autokatetrizace, proto část pacientů není schopna léčbu dodržet.
Byl vyvinut i oxybutynin pro intravezikální aplikaci s hydroxypropylcelulózou v koncentraci 5 mg/10 ml roztoku, který se aplikuje 2krát denně. Preparát byl zkoušen pouze u malého počtu pacientů, přesto představuje možnou léčebnou alternativu u pacientů s neurogenním močovým měchýřem. V současnosti se vyvíjejí systémy pro intravezikální aplikaci, malé rezervoáry, které by se měnily jednou měsíčně cystoskopicky [50].
Propiverin
Aplikace propiverinu je populární v Německu, Rakousku a v Japonsku. V USA zatím není toto léčivo registrováno.
O propiverinu bylo zatím publikováno pouze několik randomizovaných kontrolovaných studií. Inhibiční působení propiverinu na stěnu močového měchýře popsal Haruno [51]. Účinnost propiverinu byla porovnávána s oxybutyninem nebo tolterodinem. V randomizované, dvojitě slepé multicentické studii byla porovnávána účinnost léčby propiverinem podávaným 2krát denně v dávce 15 mg s tolterodinem IR v dávce 2 mg 2krát denně. Prokázalo se, že propiverin je stejně účinný jako tolterodin, vykazuje stejnou snášenlivost a stejně zlepšuje kvalitu života [52]. Při porovnání s oxybutyninem byl při léčbě propiverinem zaznamenán nižší výskyt suchosti v ústech a obtíže měly nižší intenzitu. Propiverin zlepšuje urodynamické parametry stejně účinně jako oxybutynin (včetně kapacity močového měchýře, prvního nucení na močení) [53].
Při léčbě propiverinem se vyskytuje sucho v ústech u 37 % pacientek oproti 8 % žen, které dostávaly placebo.
Farmakologické vlastnosti
Propiverin je terciární aminové syntetické parasympatolytikum působící jako antagonista acetylcholinu na muskarinových receptorech, působí i jako blokátor kalciového kanálu, působí i spasmolyticky na hladkou svalovinu močového měchýře [54].
Průměrná absolutní biologická dostupnost propiverinu je asi 40 %. Po perorálním podání se rychle a prakticky úplně absorbuje, maximálních plazmatických koncentrací dosahuje v průměru za 2,3 hod. Propiverin podléhá vysokému first-pass metabolismu a má 3 aktivní metabolity, které vykazují účinek blokátorů vápníkového kanálu. Hlavní aktivní metabolit N-oxid propiverinu se v plazmě nachází ve vyšší koncentraci než výchozí látka. Dva další aktivní metabolity se v plazmě nacházejí v nízkých množstvích. Při opakovaném (2–3krát denně) podávání jednotlivých dávek je dosaženo rovnovážného stavu po 4 dnech. Ani při dlouhodobé terapii nedochází ke kumulaci. Vazba na bílkoviny plazmy je cca 95 %, pro hlavní metabolit cca 60 %. Propiverin je vylučován močí především v metabolizované formě. Průměrný poločas eliminace je 20 hodin.
Trospium chlorid
Trospium chlorid je kvarterní amoniová sloučenina, která se v Evropě používá pro léčbu OAB více než 20 let, FDA povolila užívání trospia v USA teprve nedávno.
Oproti jiným léčivům má některé teoretické výhody: neproniká hematoencefalickou bariérou (nemá vedlejší efekt na CNS a neovlivňuje kognitivní funkce). Neváže se na bílkoviny a není metabolizován systémem cytochromu P-450, tím se snižuje riziko možných lékových interakcí.
V klinických studiích byl trospium chlorid účinnější než placebo v redukci frekvence mikcí, ve snížení intenzity nutkání k močení, ve snížení počtu epizod inkontinence. Trospium chlorid zvyšoval objem vyloučené moče o 80–140 ml. Ve srovnávacích klinických studiích se trospium chlorid ukázal minimálně stejně účinný jako oxybutynin a tolterodin [55–58].
Nežádoucí účinky trospium chloridu vycházejí z jeho anticholinergního působení, ve většině případů nevedly k nutnosti ukončení léčby. V třítýdenní klinické studii zahrnující 208 pacientů a srovnávající skupinu pacientů užívajících 20 mg trospia podávaného dvakrát denně a skupinu pacientů užívajících placebo bylo hlášeno srovnatelné celkové množství nežádoucích příhod (68 v. 62 %). Skupiny se statisticky významně lišily ve frekvenci pacientů stěžujících si na pocit sucha v ústech (43 v. 18 pacientů dostávajících placebo) [58]. V rozsáhlejší randomizované klinické studii s 523 pacienty trvající 12 týdnů, v níž byli pacienti též rozděleni do skupin, jimž bylo podáváno trospium (20 mg 2krát denně) a placebo, byla zaznamenána incidence suchosti v ústech u 21,8 % pacientů léčených trospiem oproti 6,5 % u pacientů užívajících placebo. V této studii bylo pozorováno též zvýšené procento pacientů trpících zácpou (9,5 % v 3,8 %) a bolestmi hlavy (6,5 % v 4,6 %) [55].
Farmakologické vlastnosti
Trospium chlorid je kompetitivní antagonista acetylcholinu s vysokou vazebnou aktivitou k muskarinovým receptorům M1, M2 a M3. K nikotinovým a adrenergním receptorům se neváže. Ve studiích in vitro vykazoval trospium chlorid ve srovnání s oxybutyninem, tolterodinem, darifenacinem a solifenacinem vyšší vazebnou aktivitu k M-receptorům (tab. 1) [15–18]. Na účinnosti in vivo se však podílí mnoho dalších faktorů.
Jako kvarterní amoniová látka je trospium relativně hydrofilní, a proto je jeho vstřebávání pomalé a neúplné. Jeho biologická dostupnost se nejčastěji pohybovala okolo 10 %. Maximální plazmatické koncentrace bylo dosaženo 5–6 hodin po jednorázovém podání 20 mg látky. Vstřebávání trospia je závislé na denní době (33% snížení AUC večer oproti ranním hodnotám) a na příjmu potravy (tučná strava způsobila 70–80% snížení hodnot AUC). Trospium se váže na plazmatické bílkoviny z 50–85 %. Jaterní metabolismus probíhá na cytochromu CYP2D6, nicméně je poměrně nízký. Převážná část vstřebané frakce trospia je vylučována ledvinami; v moči bylo zjištěno zhruba 60 % ve formě nezměněného trospia, asi 40 % bylo tvořeno metabolity [59].
Tolterodin s rychlým uvolňováním
Již dřívější studie prokázaly účinnost tolterodinu IR při léčbě příznaků hyperaktivního močového měchýře. Také prokázaly, že má výrazné nežádoucí účinky. Účinnost a bezpečnost tolterodinu IR podávaného 2krát denně v dávce 2 mg byla testována v několika randomizovaných dvojitě slepých studiích kontrolovaných placebem.
Ve studii Millarda a kol. byl podáván tolterodin v dávce 1 mg či 2 mg a srovnáván s placebem. Silnější dávka tolterodinu snížila počet mikcí z 11 na 9 (p < 0,05), zvýšila objem vylučované moče z průměrných 155 ml na 190 ml (p < 0,05), avšak statisticky nevýznamně snížila epizody inkontinence [60]. Zhodnocení výsledků 4 studií s celkem 1120 pacienty přineslo podobné závěry: tolterodin v dávce 4 mg/den byl stejně účinný jako oxybutynin v dávce 15 mg/den ve snížení inkontinenčních epizod, ve zvýšení objemu moče, ve snížení frekvence mikcí a v ovlivnění dalších parametrů [61]. V jiné metaanalýze ze 4 krátkodobých studií byl tolterodin IR stejně účinný jako oxybutynin IR ve snížení počtu mikcí za den, nicméně oxybutynin účinněji snižoval počet inkontinenčních epizod během dne (rozdíl průměrů 0,41; 95% CI: 0,04–0,77) a zvyšoval průměrný objem vyloučené moče (rozdíl průměrů 8,24 ml; 95% CI: 3,38–14,19). Tolterodin byl však v klinických studiích lépe tolerován. Na suchost v ústech při léčbě tolterodinem ve srovnání s léčbou oxybutyninem si stěžovala polovina pacientů (RR = 0,54; 95% CI: 0,48–0,61). Také riziko odstoupení ze studie kvůli nežádoucím účinkům bylo výrazně nižší při léčbě tolterodinem (RR = 0,63; 95% CI: 0,46–0,88). Počet nežádoucích příhod při léčbě tolterodinem byl podobný jako při podávání placeba [62].
Farmakologické vlastnosti
Tolterodin je in vivo kompetitivní antagonista muskarinových receptorů s vyšší selektivitou pro močový měchýř než pro receptory slinných žláz. Účinek léčby lze očekávat do 4 týdnů. Tolterodin se rychle vstřebává a rychle dosahuje maximálních sérových koncentrací za 30 až 120 minut po podání [63–65]. Tolterodin se metabolizuje převážně polymorfním enzymem CYP2D6 na 5-hydroxymethyl metabolit, který vykazuje farmakologický profil podobný profilu mateřské látky. Přibližně 7 % populace má defekt CYP2D6. U těchto lidí se uplatňuje CYP3A4 a dealkylace tolterodinu na N-dealkylovaný tolterodin. Bezpečnost i tolerabilita léku je pro obě skupiny pacientů stejná, a není nutné upravovat dávkování. Jídlo neovlivňuje biologickou dostupnost. U osob s cirhózou jater byla zjištěna přibližně dvojnásobně vyšší expozice volnému tolterodinu a 5-hydroxymethyl metabolitu. Nežádoucí účinky po podání tolterodinu jsou nejspíše následkem vysokých sérových koncentrací po podání tolterodinu IR [66]. Snížení maximálních sérových koncentrací zlepšuje snášenlivost léku. Podobně jako při podávání oxybutyninu mnoho pacientů přeruší léčbu v průběhu 6 měsíců pro nežádoucí účinky. Výše uvedené důvody vedly k vývoji léku s postupným uvolňováním. Preparát IR již není na našem trhu k dispozici.
Tolterodin s prodlouženým uvolňováním
Tolterodin s prodlouženým uvolňováním (ER) je výhodnější než IR. Ve velké dvojitě slepé studii kontrolované placebem byla porovnána účinnost tolterodinu ER podávaného v dávce 4 mg 1krát denně s tolterodinem podávaným v dávce 2 mg 2krát denně. Oba dva režimy prokázaly signifikantní zlepšení příznaků hyperaktivního močového měchýře oproti placebu. Tolterodin ER redukoval týdenní počet epizod inkontinence o 11,8 (p = 0,0001 oproti placebu), tolterodin IR redukoval týdenní počet epizod inkontinence o 10,6 (p = 0,0005 oproti placebu). Byla prokázána i redukce celkového počtu mikcí. Suchost v ústech se ve skupině ER vyskytovala u 23 % žen, ve skupině IR u 30 % žen a ve skupině placeba u 8 % žen. Tolterodin s postupným uvolňováním vykazuje vyšší účinnost a nižší výskyt nežádoucích účinků [66].
V jiné studii byl porovnán efekt léčby oxybutyninem ER a tolterodinem ER [67]. U obou skupin léčených žen byla prokázána stejná efektivita léčby, ale oxybutynin byl účinnější při redukci celkového počtu mikcí. Suchost v ústech se vyskytla u 29 % žen léčených oxybutyninem ER a u 22,3 % léčených tolterodinem ER (p = 0,02). Ostatní typické nežádoucí účinky se nelišily mezi oběma skupinami žen. Snížený výskyt suchosti v ústech se vysvětluje postupnějším uvolňováním tolterodinu ER [66].
Farmakologické vlastnosti
Většina farmakologických charakteristik tolterodinu byla popsána výše. Tolterodin s pomalým uvolňováním se liší absorpcí léčiva. Tolterodin ER a jeho 5-hydroxymethyl derivát dosahují oproti formě IR nižších sérových koncentrací, maximálních sérových koncentrací je dosaženo až za 2–6 hodin po podání léčiva. Minimální koncentrace je u formy ER 1,5krát vyšší než u formy IR. V rozmezí terapeutických dávek je farmakokinetika lineární. Poločas tolterodinu podaného ve formě tobolek s prodlouženým uvolňováním je přibližně 6 hodin a poločas jeho 5-hydroxymethyl derivátu je stejný. Rovnovážného stavu je dosaženo do 4 dnů od zahájení podávání přípravku. Jídlo neovlivňuje biologickou dostupnost. U osob s cirhózou jater byla zjištěna přibližně dvojnásobně vyšší expozice volnému tolterodinu a 5-hydroxymethyl metabolitu [64].
Solifenacin
Solifenacin je kompetitivní antagonista cholinergních receptorů M2 a M3 s vyšší selektivitou k močovému měchýři než k slinným žlázám. Je účinný při léčbě všech symptomů hyperaktivního močového měchýře, léčbu solifenacinem provází nižší výskyt suchosti v ústech.
V II. fázi klinického zkoušení byla porovnána aplikace 5 mg a 10 mg solifenacinu s tolterodinem podávaným v dávce 2 mg 2krát denně v randomizované, dvojitě slepé multicentrické studii kontrolované placebem [68]. Hodnoceno bylo 1033 pacientů. Aplikace 5 mg a 10 mg solifenacinu, nikoli však tolterodinu, v této studii výrazně snížila denní výskyt epizod urgence a urgentní inkontinence (p < 0,001). Solifenacin v dávce 5 mg a 10 mg redukoval oproti placebu urgentní epizody o 2,85, resp. o 3,07 (p < 0,001). Ve srovnání s placebem snížil tolterodin i solifenacin počet mikcí za den, objem moče byl zvýšen na konci studie průměrně o 31 ml při podávání 5 mg solifenacinu denně, o 35 ml při podávání 10 mg solifenacinu denně a o 20 ml při léčbě tolterodinem v dávce 2 mg/den.
Ve studii Cardozo a kol. snižoval solifenacin počet mikcí (5 mg o 2,37, p = 0,0018; 10 mg o 2,81, p = 0,0001), snižoval výskyt urgence (5 mg o 2,84, p = 0,003; 10 mg o 2,9, p = 0,002), snižoval nykturii (5 mg nesignifikantně; 10 mg o 0,71, p = 0,036) a zvyšoval objem moče při mikci [69].
Studie STAR, do níž bylo zařazeno 1355 pacientů ze 17 zemí, prokázala celkem u 74 % pacientů po léčbě solifenacinem minimálně 50% snížení počtu epizod inkontinence, přičemž 59 % léčených pacientů bylo plně kontinentních [70].
Nižší výskyt nežádoucích účinků při léčbě solifenacinem a vyšší snášenlivost léčby se vysvětluje jeho vyšší selektivitou pro tkáň močového měchýře. Při studiu afinity oxybutyninu a solifenacinu ke tkáni močového měchýře u myši se prokázala jejich stejná vazebná kapacita, ale solifenacin měl nižší vazebnou schopnost vůči tkáni slinné žlázy. Solifenacin má vyšší selektivitu k receptorům M3 než k receptorům M2 a má vyšší selektivitu ke tkáni močového měchýře než ke slinné žláze [17]. V klinických studiích bylo sucho v ústech nejčastějším nežádoucím účinkem, objevilo se u 11 % pacientů léčených dávkou 5 mg solifenacinu denně, u 22 % pacientů léčených dávkou 10 mg solifenacinu denně a u 4 % pacientů užívajících placebo [71]. Dalšími nežádoucími příhodami vedoucími k ukončení účasti ve studii byla obstipace (5,4 % pacientů léčených solifenacinem v dávce 5 mg/den, 8,5 % pacientů s dávkou 10 mg/den a 1,9 % pacientů užívajících placebo) a rozostřené vidění (3,8 % v 5,9 % v 2,5 % užívajících placebo) [68, 69].
Farmakologické vlastnosti
Po užití tablet je maximální plazmatické koncentrace dosaženo za 3–8 hod., absolutní biologická dostupnost je přibližně 90 %. Příjem potravy nemá na cmax a AUC žádný vliv. Solifenacin se přibližně z 98 % váže na plazmatické proteiny, především na kyselý a1-glykoprotein. Významně se metabolizuje v játrech, především systémem cytochromu CYP3A4. Systémová clearance je přibližně 9,5 l/hod., biologický poločas eliminace 45–68 hodin.
Darifenacin ER
Darifenacin je vysoce selektivní antagonista receptoru M3 a má k němu 5krát vyšší afinitu než k receptoru M1.
Při analýze tří 12týdenních dvojitě slepých randomizovaných studií kontrolovaných placebem bylo při aplikaci 7,5 mg a 15 mg darifenacinu prokázáno signifikantní zlepšení oproti placebu. Výskyt epizod inkontinence byl snížen o 8,8 (68,4 %) při léčbě dávkou 7,5 mg darifenacinu denně a o 10,6 (76,8 %) při podávání dávky 15 mg/den (p < 0,01). Darifenacin byl účinnější než placebo i v dalších parametrech – snižoval frekvenci mikcí, zvyšoval objem vyloučené moče, snižoval intenzitu nutkání k mikci [72]. Při další podrobné analýze se prokázala stejná účinnost u starších pacientů nad 65 let a zároveň byla prokázána i redukce nykturie [73, 74].
V klinických studiích se při léčbě dávkou 7,5 mg vyskytla suchost v ústech u 20 % pacientů, při podávání dávky 15 mg byla suchost v ústech zaznamenána u 35 % pacientů. Oproti placebu byla ve studiích pozorována častěji také zácpa (14,8 % a 21 % po léčbě dávkami 7,5 mg a 15 mg darifenacinu denně, zatímco v 5,4 % při podávání placeba). Účinky na CNS a kardiovaskulární aparát byly srovnatelné s placebem [72].
Farmakologické vlastnosti
Velmi dobrá snášenlivost preparátu se dá vysvětlit vysokou tkáňovou selektivitou. Darifenacin má 9krát vyšší selektivitu ke tkáni močového měchýře než ke slinné žláze [26].
Vzhledem k first-pass efektu je biologická dostupnost darifenacinu v ustáleném stavu přibližně 15% po podání denní dávky 7,5 mg a 19% po podání denní dávky 15 mg. Maximálních hladin v plazmě je dosaženo přibližně za 7 hodin po podání tablet s prodlouženým uvolňováním a rovnovážného stavu je dosaženo šestý den aplikace. Potrava nemá vliv na farmakokinetiku darifenacinu. Darifenacin je lipofilní povahy a z 98 % se váže na plazmatické bílkoviny (hlavně na kyselý a1-glykoprotein). Distribuční objem v ustáleném stavu je 163 litrů. Darifenacin je významně metabolizován cytochromy CYP3A4 a CYP2D6 v játrech a CYP3A4 ve stěně střevní. Látka se vylučuje ze 60 % močí a ze 40 % do stolice. Eliminační poločas je zhruba 3 hodiny.
Pro umožnění jednodenní aplikace byla aktivní látka obklopena nerozpustnou matrix, která zabezpečuje postupné uvolňování. Darifenacin se aplikuje 1krát denně s tekutinou, může být aplikován nalačno i po jídle. Jeho používání není doporučeno u pacientů s výrazným zhoršením jaterních funkcí.
Závěr
V poslední době výrazně sílí snaha o zvýšení snášenlivosti léčby anticholinergiky. Do praxe se zavádějí nové látky a modifikují se cesty aplikace stávajících léků. Příkladem modifikace v aplikaci je tolterodin a oxybutynin. Obě látky se původně aplikovaly 2–3krát denně a obě látky existují ve formě s prodlouženým uvolňováním, která umožňuje aplikaci 1krát denně a snižuje incidenci nežádoucích účinků (oxybutynin s postupným uvolňováním bohužel není v ČR k dispozici). Bohužel, na našem trhu není k dispozici ani oxybutynin k transdermální aplikaci, který se aplikuje 2krát týdně a opět výrazně zvyšuje pohodlí při aplikaci a redukuje výskyt nežádoucích účinků, hlavně suchosti v ústech. Redukce nežádoucích účinků bylo dosaženo snížením hladiny metabolitu oxybutyninu (N-DEO) v plazmě, který je odpovědný za většinu nežádoucích účinků.
Další látka, trospium chlorid, se vyznačuje nižším výskytem nežádoucích účinků díky tomu, že neproniká hematoencefalickou bariérou. Dalším rozšířením léčebných možností je zavedení nových selektivních léčiv solifenacinu a darifenacinu do klinické praxe. Všechny tyto látky mají podobnou účinnost jako oxybutynin. Léčba těmito látkami ve srovnání s léčbou oxybutyninem a tolterodinem s prodlouženým uvolňováním je však provázena výrazně nižším výskytem nežádoucích účinků, jako je suchost v ústech. Pochopení role jednotlivých subtypů muskarinových receptorů a jejich úlohy při ovlivnění funkce močového měchýře povede k rozšíření možností léčby příznaků hyperaktivního močového měchýře.
Seznam použité literatury
- [1] Chess-Williams R. Muscarinic receptors of the urinary bladder: detrusor, urothelial and prejunctional. Auton Autacoid Pharmacol 2002; 22: 133–145.
- [2] Abrams P, Cardozo L, Fall M, et al. Standardisation Sub-committee of the International Continence Society. The standardisation of terminology of lower urinary tract function: report from the Standardisation Sub-committee of the International Continence Society. Neurourol Urodyn 2002; 21: 167–178.
- [3] Bates P. Fourth report on the standardisation of terminology of lower urinary tract function. Terminology related to neuromuscular dysfunction of the lower urinary tract. Produced by the International Continence Society. Br J Urol 1981; 53: 333–335.
- [4] Walters MD, Taylor S, Schoenfeld LS. Psychosexual study of women with detrusor instability. Obstet Gynecol 1990; 75: 22–26.
- [5] Norton KR, Bhat AV, Stanton SL. Psychiatric aspects of urinary incontinence in women attending an outpatient urodynamic clinic. BMJ 1990; 301: 271–272.
- [6] Nergardh A, Boreus LO, Holme T. The inhibitory effect of coliendotoxin on alpha-adrenergic receptor functions in the lower urinary tract. An in vitro study in cats. Scand J Urol Nephrol 1977; 11: 219–224.
- [7] Martan A, Mašata J, Švabík K. Inkontinence moči u žen a její medikamentózní léčba. Praha, Maxdorf, 2005.
- [8] Schwantes U, Topfmeier P. Importance of pharmacological and physicochemical properties for tolerance of antimuscarinic drugs in the treatment of detrusor instability and detrusor hyperreflexia-chances for improvement of therapy. Int J Clin Pharmacol Ther 1999; 37: 209–218.
- [9] Yoshimura N, Chancellor MB. Current and future pharmacological treatment for overactive bladder. J Urol 2002; 168: 1897–1913.
- [10] Schmidt T, Widmer R, Pfeiffer A, Kaess H. Effect of the quaternary ammonium compound trospium chloride on 24 hour jejunal motility in healthy subjects.Gut 1994; 35: 27–33.
- [11] Nilvebrant L. The mechanism of action of tolterodine. Rev Contemp Pharmacother 2000; 11: 13–27.
- [12] Caulfield MP, Birdsall NJ. International Union of Pharmacology. XVII. Classification of muscarinic acetylcholine receptors. Pharmacol Rev 1998; 50: 279–290.
- [13] Chapple CR, Yamanishi T, Chess-Williams R. Muscarinic receptor subtypes and management of the overactive bladder. Urology 2002; 60 (Suppl. 1): 82–88.
- [14] Andersson KE. The overactive bladder: pharmacologic basis of drug treatment. Urology 1997; 50 (6A Suppl): 74–84.
- [15] Nilvebrant L, Andersson KE, Gillberg PE, et al. Tolterodine – a new bladder-selective antimuscarinic agent. Eur J Pharmacol 1997; 127: 195–207.
- [16] Gillberg PG, Sundquist S, Nilvebrant L. Comparison of the in vitro and in vivo profiles of tolterodine with those of subtype-selective muscarinic receptor antagonists. Eur J Pharmacol 1998; 349: 285–292.
- [17] Ikeda K, Kobayashi S, Suzuki M, et al. M(3) receptor antagonism by the novel antimuscarinic agent solifenacin in the urinary bladder and salivary gland. Naunyn Schmiedebergs Arch Pharmacol 2002; 366: 97–103.
- [18] Napier CM, Gupta P. Darifenacin is selective for the human recombinant M3 receptor subtype. Neurourol Urodyn 2002; 21: A445.
- [19] Barras M, Coste A, Eon MT, et al. Pharmacological characterization of muscarinic receptors implicated in rabbit detrusor muscle contraction and activation of inositol phospholipid hydrolysis in rabbit detrusor and parotid gland. Fundam Clin Pharmacol 1999; 13: 562–570.
- [20] Sawyer GW, Lambrecht G, Ehlert FJ. Functional role of muscarinic M(2) receptors in alpha,beta-methylene ATP induced, neurogenic contractions in guinea-pig ileum. Br J Pharmacol 2000; 129: 1458–1464.
- [21] Pak RW, Petrou SP, Staskin DR. Trospium chloride: a quaternary amine with unique pharmacologic properties. Curr Urol Rep 2003; 4: 436–440.
- [22] Kobayashi S, Ikeda K, Miyata K. Comparison of in vitro selectivity profiles of solifenacin succinate (YM905) and current antimuscarinic drugs in bladder and salivary glands: a Ca2+ mobilization study in monkey cells. Life Sci 2004; 74: 843–853.
- [23] Hatanaka T, Ukai M, Ohtake A, et al. in vitro tissue selectivity profile of solifenacin succinate (YM905) for urinary bladder over salivary gland in rats and monkeys [abstract 312]. Presented at the International Continence Society Meeting. Florence, Italy: 2003 Oct 5–9.
- [24] Ohtake A, Hatanaka T, Ikeda K, et al. In vivo bladder selective profiIe of solifenacin succinate (YM905) over salivary gland in mice and rats. Presented at the International Continence Society Meeting [Abstract 297]. Florence, Italy: October 5–9, 2003.
- [25] Kershen RT, Hsieh M. Preview of new drugs for overactive bladder and incontinence: darifenacin, solifenacin, trospium, and duloxetine. Curr Urol Rep 2004; 5: 359–367.
- [26] Newgreen OT, Anderson OW, Carter AJ, et al. Darifenacin: a novel bladder-selective agent for the treatment of urge incontinence. Neurourol Urodyn 1995; 14: 557.
- [27] Madersbacher H, Knoll M. Intravesical application of oxybutynine: mode of action in controlling detrusor hyperreflexia. Preliminary results. Eur Urol 1995; 28: 340–344.
- [28] Walter P, Grosse J, Bihr AM, et al. Bioavailability of trospium chloride after intravesical instillation in patients with neurogenic lower urinary tract dysfunction: A pilot study. Neurourol Urodyn 1999; 18: 447–453.
- [29] Frohlich G, Burmeister S, Wiedemann A, Bulitta M. Intravesical instillation of trospium chloride, oxybutynin and verapamil for relaxation of the bladder detrusor muscle. A placebo controlled, randomized clinical test. Arzneimittelforschung 1998; 48: 486–491.
- [30] Riva O, Casolati E. Oxybutynin chloride in the treatment of female idiopathic bladder instability: results from double blind treatment. Clin Exp Obstet Gynecol 1984; 11: 37–42.
- [31] Tapp AJ, Cardozo LD, Versi E, Cooper D. The treatment of detrusor instability in post-menopausal women with oxybutynin chloride: a double blind placebo controlled study. Br J Obstet Gynaecol 1990; 97: 521–526.
- [32] Douchamps J, Derenne F, Stockis A, et al. The pharmacokinetics of oxybutynin in man. Eur J Clin Pharmacol 1988; 35: 515–520.
- [33] Hughes KM, Lang JC, Lazare R, et al. Measurement of oxybutynin and its N-desethyl metabolite in plasma, and its application to pharmacokinetic studies in young, elderly and frail elderly volunteers. Xenobiotica 1992; 22: 859–869.
- [34] Yarker YE, Goa KL, Fitton A. Oxybutynin: a review of its pharmacodynamic and pharmacokinetic properties, and its therapeutic use in detrusor instability. Drugs Aging 1995; 6: 243–262.
- [35] Waldeck K, Larsson B, Andersson KE. Comparison of oxybutynin and its active metabolite, N-desethyl-oxybutynin, in the human detrusor and parotid gland. J Urol 1997; 157: 1093–1097.
- [36] Gupta SK, Sathyan G. Pharmacokinetics of an oral once-a-day controlled-release oxybutynin formulation compared with immediate-release oxybutynin. J Clin Pharmacol 1999; 39: 289–296.
- [37] Gleason DM, Susset J, White C, et al. Evaluation of a new once-daily formulation of oxbutynin for the treatment of urinary urge incontinence. Ditropan XL Study Group. Urology 1999; 54: 420–423.
- [38] Anderson RU, Mobley D, Blank B, et al. Once daily controlled versus immediate release oxybutynin chloride for urge urinary incontinence: OROS Oxybutynin Study Group. J Urol 1999; 161: 1809–1812.
- [39] Appell RA, Sand P, Dmochowski R, et al. Prospective randomized controlled trial of extended-release oxybutynin chloride and tolterodine tartrate in the treatment of overactive bladder: results of the OBJECT Study. Mayo Clin Proc 2001; 76: 358–363.
- [40] Zobrist RH, Quan O, Thomas HM, et al. Pharmacokinetics and metabolism of transdermal oxybutynin: in vitro and in vivo performance of a novel delivery system. Pharm Res 2003; 20: 103–109.
- [41] Zobrist RH, Schmid B, Feick A, et al. Pharmacokinetics of the R- and S-enantiomers of oxybutynin and N-desethyl-oxybutynin following oral and transdermal administration of the racemate in healthy volunteers. Pharm Res 2001; 18: 1029–1034.
- [42] Appell RA, Chancellor M, Zobrist RH, et al. Pharmacokinetics, metabolism, and saliva output during transdermal and extended-release oral oxybutynin administration in healthy subjects. Mayo Clin Proc 2003; 78: 696–702.
- [43] Davila GW, Daugherty CA, Sanders SW; Transdermal Oxybutynin Study Group. A short-term, multicenter, randomized double-blind dose titration study of the efficacy and anticholinergic side effects of transdermal compared to immediate release oral oxybutynin treatment of patients with urge urinary incontinence. J Urol 2001; 166: 140–145.
- [44] Dmochowski RR, Davila GW, Zinner NR, et al.; Transdermal Oxybutynin Study Group. Efficacy and safety of transdermal oxybutynin in patients with urge and mixed urinary incontinence. J Urol 2002; 168: 580–586.
- [45] Dmochowski RR, Sand PK, Zinner NR, et al. Transdermal Oxybutynin Study Group. Comparative efficacy and safety of transdermal oxybutynin and oral tolterodine versus placebo in previously treated patients with urge and mixed urinary incontinence. Urology 2003; 62: 237–242.
- [46] Radziszewski P, Borkowski A. Therapeutic effects of intrarectal administration of oxybutynin. Wiad Lek 2002; 55: 691–698.
- [47] Collas D, Malone-Lee J. The pharmacokinetic properties of rectal oxybutynin: a possible altemative to intravesical administration. In: ICS 27th Annual Meeting; 1997 Sep 23–26; Yokohama, Japan: Neurourology and Urodynamics; 1997: 346.
- [48] Buyse G, Waldeck K, Verpoorten C, et al. Intravesical oxybutynin for neurogenic bladder dysfunction: less systemic side effects due to reduced first pass metabolism. J Urol 1998; 160: 892–896.
- [49] Weese DL, Roskamp DA, Leach GE, Zimmern PE. Intravesical oxybutynin chloride: experience with 42 patients. Urology 1993; 41: 527–530.
- [50] Cannon TW, Chancellor MB. Pharmacotherapy of the overactive bladder and advances in drug delivery. Clin Obstet Gynecol 2002; 45: 205–217.
- [51] Haruno A. Inhibitory effects of propiverine hydrochloride on the agonist-induced or spontaneous contractions of various isolated muscle preparations. Drug Res 1992; 42: 815.
- [52] Junemann KP, Halaska M, Rittstein T, et al. Propiverine versus tolterodine: efficacy and tolerability in patients with overactive bladder. Eur Urol 2005; 48: 478–482.
- [53] Madersbacher H, Halaska M, Voigt R, et al. A placebo-controlled, multicentre study comparing the tolerability and efficacy of propiverine and oxybutynin in patients with urgency and urge incontinence. BJU Int 1999; 84: 646–651.
- [54] Madersbacher H, Murtz G. Efficacy, tolerability and safety profile of propiverine in the treatment of the overactive bladder (non – neurogenic and neurogenic). Word J Urol 2001; 19: 324–335.
- [55] Zinner N, Gittelman M, Harris R, et al. Trospium chloride improves overactive bladder symptoms: a multicenter phase III trial. J Urol 2004; 171: 2311–2315.
- [56] Madersbacher H, Stohrer M, Richter R, et al. Trospium chloride versus oxybutynin: a randomized, double-blind, multicentre trial in the treatment of detrusor hyper-reflexia. Br J Urol 1995; 75: 452–456.
- [57] Junemann KP, Al-Shukri S. Efficacy and tolerability of trospium chloride and tolterodine in 234 patients with urge syndrome: a double-blind. placebo-controlled, multicentre, clinical trial (abstr.) Neurourol Urodyn 2000; 19: 488–490.
- [58] Cardozo L, Chapple CR, Toozs-Hobson P, et al. Efficacy of trospium chloride in patients with detrusor instability: a placebo-controlled, randomized, double-blind, multicentre clinical trial. BJU Int 2000; 85: 659–664.
- [59] Doroshyenko O, Jetter A, Odenthal KP, et al. Clinical Pharmacokinetics of Trospium Chloride. Clin Pharmacokinet 2005; 44: 701–720.
- [60] Millard R, Tuttle J, Moore K, et al. Clinical efficacy and safety of tolterodine compared to placebo in detrusor overactivity. J Urol 1999; 161: 1551–1555.
- [61] Appell RA. Clinical efficacy and safety of tolterodine in the treatment of overactive bladder: a pooled analysis. Urology 1997; 50 (6A Suppl): 90–96.
- [62] Harvey MA, Baker K, Wells GA. Tolterodine versus oxybutynin in the treatment of urge urinary incontinence: a meta-analysis. Am J Obstet Gynecol 2001; 185: 56–61.
- [63] Guay OR. Clinical pharmacokinetics of drugs used to treat urge incontinence. Clin Pharmacokinet 2003; 42: 1243–1285.
- [64] Olsson B, Szamosi J. Multiple dose pharmacokinetics of a new once daily extended release tolterodine formulation versus immediate release tolterodine. Clin Pharmacokinet 2001; 40: 227–235.
- [65] Brynne N, Stahl MM, Hallen B, et al. Pharmacokinetics and pharmacodynamics of tolterodine in man: a new drug for the treatment of urinary bladder overactivity. Int J Clin Pharmacol Ther 1997; 35: 287–295.
- [66] Van Kerrebroeck P, Kreder K, Jonas U, et al. Tolterodine once-daily: superior efficacy and tolerability in the treatment of the overactive bladder. Urology 2001; 57: 414–421.
- [67] Diokno AC, Appell RA, Sand PK, et al.; OPERA Study Group. Prospective, randomized, double-blind study of the efficacy and tolerability of the extended-release formulations of oxybutynin and tolterodine for overactive bladder: results of the OPERA trial. Mayo Clin Proc 2003; 78: 687–695.
- [68] Chapple CR, Rechberger T, Al-Shukri S, et al. Randomized, double-blind placebo- and tolterodine-controlled trial of the once-daily antimuscarinic agent solifenacin in patients with symptomatic overactive bladder. BJU Int 2004; 93: 303–310.
- [69] Cardozo L, Lisec M, Millard R, et al. Randomized, double-blind placebo controlled trial of the once daily antimuscarinic agent solifenacin succinate in patients with overactive bladder. J Urol 2004; 172 (5 Pt 1): 1919–1924.
- [70] Chapple CR, Martinez-Garcia R, Selvaggi L, et al.; for the STAR study group. A comparison of the efficacy and tolerability of solifenacin succinate and extended release tolterodine at treating overactive bladder syndrome: results of the STAR trial. Eur Urol 2005; 48: 464–470.
- [71] Solifenacin – SPC. MV-AISLP 1.2007.
- [72] Chapple C, Steers W, Norton P, et al. A pooled analysis of three phase III studies to investigate the efficacy, tolerability and safety of darifenacin, a muscarinic M3 selective receptor antagonist, in the treatment of overactive bladder. BJU Int 2005; 95: 993–1001.
- [73] Foote JE. Darifenacin a highly M3 selective muscarinic receptor antagonist is effective and well tolerated by elderly patients with overactive bladder. J Am Geriatr Soc 2004; 52 (Suppl. 4): SI26.
- [74] Khullar V. Darifenacin, an M3 selective receptor antagonist, reduces the frequency of nocturnal awakening, an important symptom of overactive bladder. J Urol 2004; 171 (Suppl. 4): 131.