Biktarvy – přípravek s novým integrázovým inhibitorem v jednotabletovém režimu v léčbě infekce HIV
Souhrn:
Sedláček D, Hofman S. Biktarvy – přípravek s novým integrázovým inhibitorem v jednotabletovém režimu v léčbě infekce HIV. Remedia 2019; 29: 277–283.
Biktegravir (BIC) je nový inhibitor integrázy HIV 1 i HIV 2 druhé generace. V únoru 2018 byl schválen americkým Úřadem pro kontrolu potravin a léčiv (FDA) v kombinaci s emtricitabinem a tenofovir alafenamidem (BIC/F/TAF – 50/200/25 mg, Biktarvy®) pro léčbu dospělých osob s infekcí HIV 1. Podává se jedna tableta jednou denně. Jeho účinnost, bezpečnost, snášenlivost a lékové interakce byly hodnoceny ve čtyřech (GS-US-380-1489, 1490, 1844 a 1878) studiích s použitím srovnatelného komparátoru – dolutegraviru či inhibitoru proteázy u předléčených pacientů. Indikací BIC/F/TAF je infekce HIV 1 u osob dosud neléčených nebo u dříve léčených bez známek selhání před-chozího režimu a bez prokázané rezistence na některou komponentu. Ve sledovaném období 96 týdnů nebyly zjištěny rezistence na BIC. Další studie, včetně poregistračních, probíhají
Summary:
Sedlacek D, Hofman S. Biktarvy – a product with a new integrase inhibitor, a single tablet regimen in treatment of HIV infection. Reme-dia 2019; 29: 277–283.
Bictegravir (BIC) is a novel, second generation HIV 1 and HIV 2 integrase strand transfer inhibitor. In February 2018, it has been approved by the Food and Drug Administration (FDA) in combination with emtricitabine and tenofovir alafenamide (BIC/F/TAF – 50/200/25 mg, Biktarvy®) for the treatment of adult patients with HIV 1 infection. It is administered with one tablet once daily. Its efficiency, safety, tolerability and drug interactions were elucidated in four (GS-US-380-1489, 1490, 1844 and 1878) studies using a comparator ‒ dolute-gravir, or protease inhibitor in naïve patients. The indications of BIC/F/TAF include HIV 1 infection in treatment naïve patients, or in previously treated patients with no signs of failure of the previous regimen and without a proven resistance to any component. In the period 96 weeks, no resistance to BIC was found. Other studies, including the post marketing ones, are underway.
Key words: HIV, integrase inhibitors, antiretroviral therapy, clinical studies, adverse effects, drug interactions
Úvod
Vývoj antiretrovirové terapie (antiretroviral treatment,
ART) se v posledních letech zaměřuje na bezpečnější léčiva vyšších
generací, kombinovaná zpravidla do jednotabletových režimů. Ty umožňují
vyšší adherenci k léčbě než podávání jednotlivých přípravků
ve vícetabletovém schématu. Od roku 2015 se rovněž změnil přístup
k iniciaci ART. Léčba se nyní zahajuje u každého HIV (human
immunodeficiency virus) pozitivního pacienta časně, bez ohledu na výši
imunologických a virologických parametrů. Nové doporučené postupy,
připravované předními světovými odborníky, upřednostňují léčbu inhibitory HIV‑1
integrázy v kombinaci s nukleosidovými/nukleotidovými inhibitory HIV‑1
reverzní transkriptázy (nucleoside/nucleotide reverse transcriptase inhibitors,
NRTI/NtRTI) jako metodu volby.
Novým inhibitorem HIV‑1 i HIV‑2 integrázy je
biktegravir (BIC). Spolu s emtricitabinem (F, nukleosidovým inhibitorem
HIV‑1 reverzní transkriptázy) a tenofovir alafenamidem (TAF, proléčivem
tenofoviru, nukleotidovým inhibitorem HIV‑1 reverzní transkriptázy) je
adjustován v jedné tabletě přípravku BiktarvyÒ. Každá tableta obsahuje 50 mg biktegraviru, 200 mg emtricitabinu a 25 mg tenofovir alafenamidu
(BIC/F/TAF). V loňském roce byl tento kombinovaný přípravek schválen
americkým Úřadem pro kontrolu potravin a léčiv (Food and Drug
Administration, FDA) a Evropskou lékovou agenturou (European Medicines
Agency, EMA) pro léčbu dospělých pacientů s infekcí HIV‑1 dosud neléčených
nebo pro již léčené osoby, u nichž se neprokázalo virologické selhání
(hodnota HIV-1 RNA trvale < 50 kopií/ml) ani žádné mutace
v genech kódujících příslušné enzymy (resistance associated mutation,
RAM), které by byly příčinou vzniku rezistence a následně selhání odpovědi
na jednotlivé komponenty léku. Tento kombinovaný přípravek se podává
ve formě jedné tablety jednou denně s jídlem nebo bez jídla. Schvalovací
proces vycházel především ze závěrů dvou randomizovaných, kontrolovaných
klinických studií, které prokázaly, že výsledky v souboru léčených
BIC/F/TAF nebyly horší než v souborech s adekvátními komparátory
(dolutegravir/abakavir/lamivudin nebo dolutegravir/F/TAF). Nový přípravek se
jeví jako účinný, spolehlivý a relativně bezpečný [1]. Registrace EMA umožnila
jeho registraci v členských zemích Evropské unie. V České republice se
očekává jeho zařazení do klinické praxe v průběhu letošního roku.
Teoretické podklady pro vedení ART
Díky úspěchům mnohaletého základního i aplikovaného
výzkumu je dnes infekce HIV‑1 považována za chronické léčitelné, ale zatím
nevyléčitelné onemocnění. Přes mnohé významné úspěchy epidemiologie,
patogeneze, imunologie, farmakologie a dalších oborů se dosud nepodařilo
účinně zasáhnout do metabolismu těch buněk, které mají ve svém genomu
zakódovanou provirovou DNA kódující HIV‑1. Její trvalá přítomnost
v organismu infikovaných osob je spojena s rizikem reaktivace
onemocnění, pokud z nejrůznějších důvodů není dlouhodobě zajištěna trvalá
suprese replikace HIV. U vysoce adherentních pacientů je moderní ART
schopna blokovat replikaci HIV do té míry, že jejich viremie – virová
nálož (viral load, VL) – je téměř nedetekovatelná. U takových
nemocných nedochází ve větší míře k dalšímu poškozování produkce
a funkce CD4+ T
lymfocytů. Snižuje se tak riziko morbidity a mortality na oportunní
infekce či nádory. Vysoce adherentní pacient s nedetekovatelnými VL je
také minimálně infekční. V posledních letech se v této souvislosti
hovoří o U = U (Undetectable = Untransmittable).
V praxi lze očekávat minimalizaci rizika sexuálního i vertikálního
přenosu a postupné plošné zlepšování epidemiologické situace.
Komponenty ART
Současná ART je založena na kombinaci
nukleosidových/nukleotidových inhibitorů HIV‑1 reverzní transkriptázy (např.
F/TAF), nenukleosidových inhibitorů HIV‑1 reverzní transkriptázy (např.
doravirin), inhibitorů HIV‑1 proteázy (např. darunavir), inhibitorů HIV‑1
integrázy (např. dolutegravir a biktegravir), méně na inhibitoru fúze
(enfuvirtid) a inhibitoru vstupu/chemokinového receptoru CCR5 (maravirok).
Za ART první volby se dnes, dle většiny doporučených postupů, považuje
kombinace inhibitoru HIV‑1 integrázy (integrase strand transfer inhibitor,
INSTI) s NRTI/NtRTI. Inhibitory HIV integrázy se záhy po zavedení
do klinické praxe ukázaly jako velmi nadějné a zaujaly vedoucí místo
v nových ART kombinacích díky rychlému nástupu účinku, minimu nežádoucích
účinků a dobré toleranci léčby.
Vlastnosti biktegraviru
Molekula BIC byla vyvinuta společností Gilead Sciences
a od roku 2016 je klinicky zkoušena (obr. 1).
Její molekulová hmotnost je 449 g/mol,
podobně jako jiné INSTI blokuje funkci HIV‑1 i HIV‑2 integrázy tím, že
váže kationty Mg2+ potřebné pro správnou činnost
integrázy, čímž brání zabudování provirové DNA HIV‑1 do genomu hostitele
a následné replikaci HIV‑1. Ve studiích in vitro
se zjistilo, že koncentrace vyvolávající 50% účinek (EC50)
pro BIC se nacházely v rozmezí <0,05‒6,6 nM;
EC95 pro BIC, upravená podle proteinu, byla 361 nM (0,162 μg/ml) pro HIV‑1 divokého
typu. Podobný účinek byl zjištěn u klinických izolátů HIV‑1 skupin M,
O a N i u subtypů A–G, ale také u HIV‑2.
V preklinických animálních studiích se neprokázaly mutagenní ani
kancerogenní účinky [1].
Po perorálním podání je BIC během 2‒4 hodin absorbován
(s tučným jídlem je plocha pod křivkou plazmatické koncentrace [AUC] až
o 24 % vyšší)
a ve více než 99 %
se váže na bílkoviny krevní plazmy. Hlavní cestou vylučování BIC je
jaterní metabolismus. Primárně je metabolizován enzymy CYP3A a UGT1A1
(UDP-glukuronyltransferáza). Po perorálním podání se stolicí vylučují až
v 60 % mateřská
látka a metabolity BIC. Do moči se vylučuje 35 % dávky, především glukuronidu BIC. Renální
exkrece intaktního BIC představuje asi 1 % dávky.
Biologický poločas BIC v plazmě byl stanoven na 17,3 hodiny (tab. 1) [1].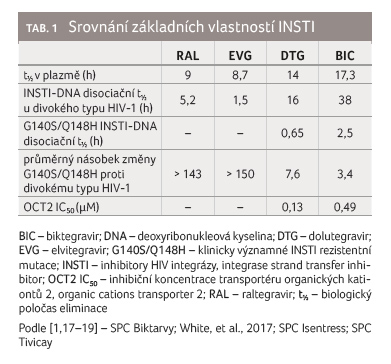
Farmakokinetika po opakovaném podání BIC je úměrná rozmezí
dávky 25‒100 mg.
Nebyly pozorovány klinicky významné rozdíly ve farmakokinetice BIC mezi
zdravými jedinci a pacienty s těžkou poruchou funkce ledvin
(odhadovaná clearance kreatininu [Clcr] < 30 ml/min) nebo se středně
těžkou poruchou funkce jater. Není dostatek informací o farmakokinetice
BIC u pacientů starších 65 let. Analýzy expozice BIC neidentifikovaly
žádné klinicky významné rozdíly s ohledem na věk, pohlaví nebo rasu
[1].
Biktegravir je substrátem CYP3A (cytochromu P450 3A)
a UGT1A1. Pokud je podán se silnými induktory CYP3A a UGT1A1 (např.
rifampicin, třezalka tečkovaná), může se podstatně snížit koncentrace BIC
a hrozí riziko rozvoje rezistence. Pokud je BIC podán se silnými
inhibitory CYP3A a UGT1A1 (např. ritonavir, kobicistat), významně se
zvyšuje jeho plazmatická koncentrace. Biktegravir je substrátem glykoproteinu P
(P‑gp) i BCRP (breast cancer resistance protein). Kombinace BIC
s inhibitory P‑gp a/nebo BCRP (např. makrolidy, cyklosporin, glekaprevir/pibrentasvir)
se proto doporučuje opatrnost při předepisování. Biktegravir inhibuje
transportér organických kationtů 2 (organic cations transporter 2, OCT2)
a mnohočetný lékový a toxinový extruzní transportér 1 (multidrug and
toxin extrusion 1, MATE1) in vitro. Zjistilo se však,
že přípravek Biktarvy lze se substráty OCT2 a MATE1 za pečlivé
kontroly renálních funkcí podávat. V játrech BIC podléhá glukuronidaci
UGT1A1 a oxidaci CYP3A4. Pouhé jedno procento BIC se vylučuje močí
nezměněno. Při renální insuficienci s Clcr < 30 ml/min a u osob
s těžkými hepatopatiemi se BIC nemá podávat [1,4].
K nejčastějším nežádoucím účinkům zjištěným
ve studiích patří cefalea (5 %),
průjem (5 %)
a nauzea (4 %)
[1]. Nežádoucí účinky týkající se poruch lipidového metabolismu a hematologických
abnormit se přisuzují většinou komedikaci NRTI/NtRTI. Vzácně se může objevit
laktátová acidóza s hepatomegalií a steatózou [5]. V takovém
případě je nutno léčbu BIC ihned ukončit. Byly rovněž pozorovány deprese, abnormální
sny, sebevražedné chování, úzkost, poruchy spánku, bolest hlavy a závratě.
Vzácně se objevila hyperbilirubinemie, angioedém, exantém, svědění kůže,
artralgie a únava. Pozdní neurologické poruchy (hypertonie, křeče,
abnormální chování) se objevují vzácně a není známo, zda jsou přechodné,
nebo trvalé. S ohledem na blízkou příbuznost BIC s DTG se tyto
nálezy musejí monitorovat především u dětí vystavených in utero
působení BIC, ale i NRTI/NtRTI [1].
U pacientů s pokročilou infekcí HIV se během
několika prvních týdnů až měsíců, podobně jako při léčbě jinými ART
kombinacemi, může rozvinout zánětlivý syndrom imunitní obnovy (immune
reconstitution inflammatory syndrome, IRIS). Jeho příčinou je asymptomatická
infekce nebo reziduální přítomnost některých původců oportunních infekcí
(nejčastěji cytomegaloviru, P. jirovecii
a mykobakterií). Důsledkem IRIS může být také autoimunitní porucha, např.
tyreotoxikóza. U nemocných s pokročilým onemocněním HIV a/nebo při
dlouhodobé expozici různým typům ART je pozorována osteopenie, osteoporóza
i osteonekróza [5].
Bylo zjištěno, že BIC zvyšuje koncentraci sérového
kreatininu v důsledku inhibice tubulární sekrece. K jeho zvýšení
v průměru o 8,8 μmol/l
došlo ve čtvrtém týdnu léčby a zůstalo stabilní až
do 48. týdne [1]. V registračních klinických studiích fáze III
s BIC nedošlo do 48. týdne, resp. do 96. týdne
u dosud neléčených pacientů k ukončení léčby z důvodu výskytu
nežádoucích renálních příhod [1,3,7,15,16].
Ve studiích GS‑US‑380‑1489 a GS‑US‑380‑1490 bylo
pozorováno zvýšení koncentrace celkového bilirubinu u 12 % dosud neléčených
pacientů. Zvýšení nebyla spojena s výskytem jaterních nežádoucích účinků
a do 96. týdne nedošlo k žádnému ukončení léčby
z důvodu výskytu nežádoucích jaterních příhod. Také u 16 dospělých
osob souběžně infikovaných HIV/HBV (virem hepatitidy B) byl bezpečnostní profil
BIC podobný jako u nemocných s monoinfekcí HIV‑1. Kontraindikací
užití BIC je hypersenzitivní reakce na některou z komponent přípravku
včetně pomocných látek, léčba rifampicinem a užívání přípravků s obsahem
třezalky tečkované [1,4,8,9].
Klinické studie pro registraci
Registraci přípravku Biktarvy předcházely dvě klinické
studie fáze III – GS‑US‑380‑1489 (n = 629) a GS‑US‑380‑1490
(n = 645) – provedené u neléčených HIV‑1 pozitivních osob (obr. 2).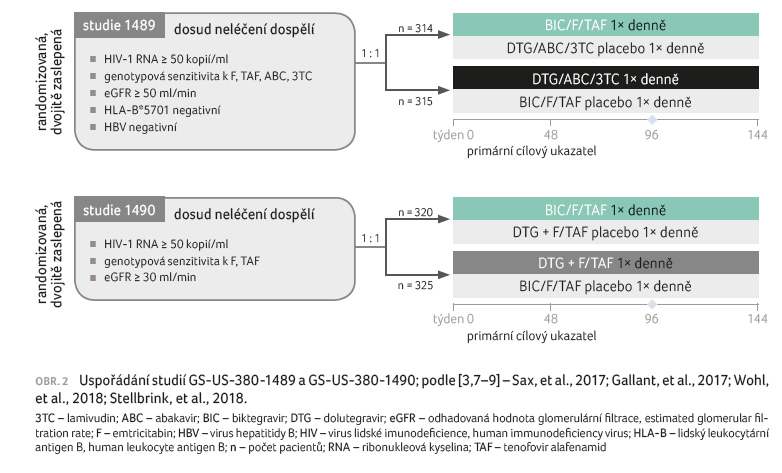
Při screeningu byly vyloučeny osoby s HIV‑1
rezistentním k NRTI užitým ve studii. V průběhu screeningu se
neprováděla genotypizace integrázy ke stanovení rezistence. Kombinace
BIC/F/TAF se po 48 a 96 týdnech vyhodnotila jako bezpečná, účinná
a srovnatelná s režimem obsahujícím DTG/ABC/3TC
(dolutegravir/abakavir/lamivudin) nebo DTG a F/TAF. Během 96 týdnů
sledování nedošlo ke vzniku rezistence na studijní medikaci. Výsledky
léčby byly podobné v obou studijních skupinách bez ohledu na věk,
pohlaví, rasu, výchozí VL a počáteční počty CD4+ T lymfocytů [3,7,15,16].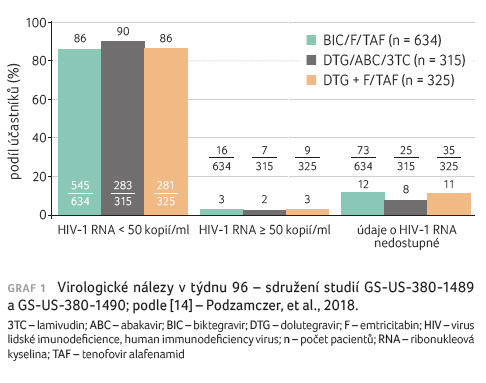
Kombinace BIC/F/TAF byla non-inferiorní v dosažení HIV‑1 RNA < 50 kopií/ml v týdnu 48 a 96 v porovnání s kombinacemi DTG/ABC/3TC a DTG/F/TAF (graf 1). Výsledky léčby byly podobné ve všech podskupinách podle věku, pohlaví, rasy, výchozí VL, výchozího počtu buněk CD4+ T lymfocytů a regionu. Ve studiích GS‑US‑380‑1489 a GS‑US‑380‑1490 bylo v týdnu 96 průměrné zvýšení počtu CD4+ buněk od výchozího stavu rovno počtu 207, 229 a 201 buněk/mm3 ve sdružených skupinách pacientů, kteří užívali BIC/F/TAF, DTG/ABC/3TC a DTG/F/TAF, v tomto pořadí. Nežádoucí příhody související s léčbou se ve studii GS‑US‑380‑1489 do 96. týdne vyskytly ve 28 % u subjektů léčených BIC/F/TAF vs. 40 % DTG/ABC/3TC (p = 0,002). Kvůli nežádoucím příhodám ukončilo léčbu do 96. týdne 0 vs. 5 subjektů. Ve studii GS‑US‑380‑
1490 BIC/F/TAF vs. DTG/F/TAF byly nežádoucí příhody celkově
srovnatelné, ale méně časté byly reakce související s hodnocenými
přípravky u pacientů léčených BIC/F/TAF 20 %
vs. 28 % (p = 0,02). K ukončení léčby z důvodu
výskytu nežádoucích účinků do týdne 96 došlo v šesti, resp. pěti
případech [8,9,15,16].
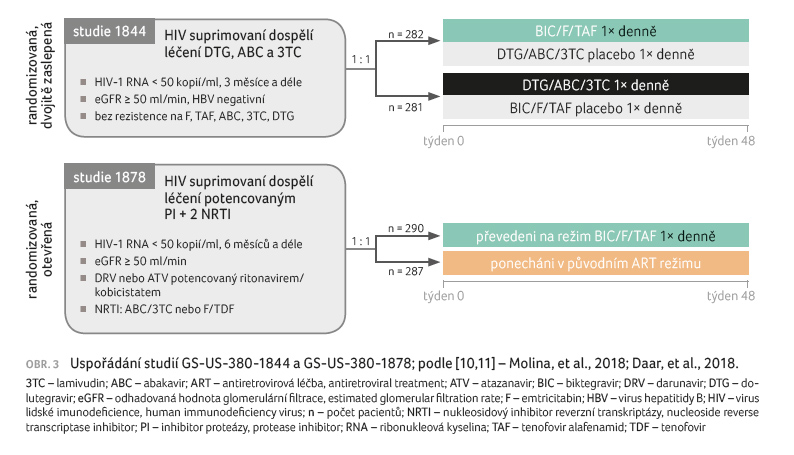
Studie GS‑US‑380‑1844 a studie GS‑US‑380‑1878
Také v dalších dvou studiích fáze III ‒ randomizované,
dvojitě zaslepené GS‑US‑380‑1844 (n = 563) a randomizované,
otevřené, aktivně kontrolované studii GS‑US‑380‑1878 (n = 577)
u účastníků s dosaženou virologickou supresí (obr. 3)
‒ byla kombinace BIC/F/TAF bezpečná, účinná a non‑inferiorní
v porovnání s léčebnými režimy založenými na inhibitoru proteázy
(PI) nebo DTG/ABC/3TC. V průběhu 48 týdnů se nevyvinula rezistence vůči
BIC/F/TAF nebo DTG/ABC/3TC
[10,11]. Výsledky terapie mezi léčebnými skupinami byly podobné napříč
podskupinami podle věku, pohlaví, rasy a regionu (graf 2).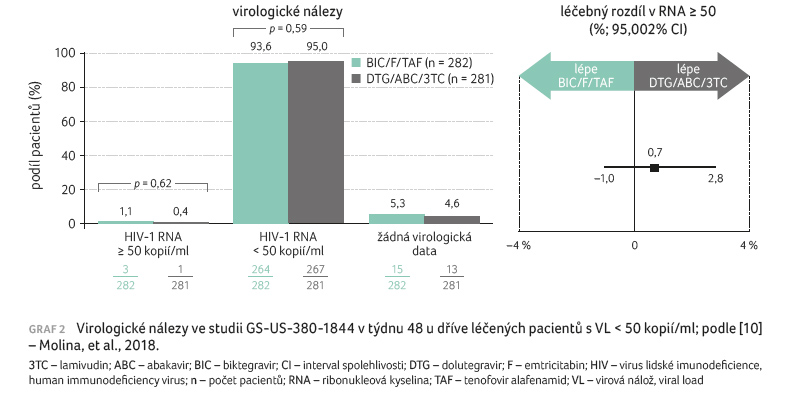
Ve studii GS‑US‑380‑1844 byla v týdnu 48 průměrná
změna počtu CD4+ T
lymfocytů v porovnání s výchozí hodnotou ‒31 buněk/mm3 u pacientů, kteří přešli na léčbu BIC/F/TAF,
a 4 buňky/mm3 u pacientů, kteří byli nadále
léčeni DTG/ABC/3TC. Ve studii GS‑US‑380‑1878 představovala
v týdnu 48 průměrná změna počtu CD4+ T
lymfocytů v porovnání s výchozí hodnotou 25 buněk/mm3 u pacientů, kteří přešli na léčbu BIC/F/TAF,
a 0 buněk/mm3 u pacientů, kteří zůstali při
léčbě výchozím režimem [1,10,11]. V současné době se již analyzují
výsledky klinických studií u suprimovaných adolescentů a dětí
starších 6 let [12].
Význam klinických výsledků pro zavedení přípravku
Biktarvy do praxe
Přípravek Biktarvy (BIC/F/TAF) obsahuje účinnou látku
bictegravirum 50 mg,
emtricitabinum 200 mg
a tenofoviri alafenamidi fumaras odpovídající 25 mg TAF. V podmínkách ČR se léčba osob
infikovaných HIV‑1 zahajuje v HIV centrech a je vedena lékařem
infektologem se zkušenostmi s léčbou infekce HIV. Přípravek se podává
jednou denně vždy ve stejnou hodinu, s jídlem nebo bez jídla, tableta
se polyká celá. V případě zvracení do jedné hodiny po příjmu
léku je nutno podat novou tabletu. Pokud byla dávka omylem vynechána
a uplynulo více než 18 hodin od doby, kdy měl být přípravek užit,
vyčká se již do doby obvyklého podání. Úprava dávkování u osob
starších 65 let, u osob s hodnotou Clcr ≥ 30 ml/min nebo s lehkou
či středně těžkou hepatopatií (podle Childova‒Pughova skóre A nebo B) není
nutná. Avšak přerušení léčby přípravkem Biktarvy (i jinými s obsahem
tenofoviru) může být u pacientů současně infikovaných HIV a HBV
spojeno s těžkou akutní exacerbací hepatitidy B [1].
Za nevhodné se považují lékové kombinace
s antacidy s obsahem hořčíku a hliníku a přípravky
s obsahem železa a vápníku. Tyto přípravky lze užít nejméně dvě
hodiny před užitím přípravku nebo dvě hodiny po něm. Další lékové
interakce jednotlivých komponent přípravku lze vyhledat např. na webových
stránkách Liverpoolské univerzity [13].
Přestože se v souboru více než 1 000 těhotných žen exponovaných přípravku Biktarvy
během těhotenství nenaznačil malformační potenciál ani toxické účinky,
doporučuje se podávat Biktarvy během těhotenství pouze v případě, že
potenciální přínos převyšuje potenciální riziko. Kojení se u HIV
pozitivních žen nedoporučuje.
Při předávkování Biktarvy neexistuje specifické antidotum
a vzhledem k vazbě na plazmatické bílkoviny nelze využít ani
dialyzační metody. Pacient je hospitalizován, monitorují se jeho základní
životní funkce a klinický stav [1,4].
Biktegravir a rezistence
Citlivost BIC byla testována na 64 klinických izolátech
rezistentních na INSTI (20 s jednou substitucí a 44 se dvěma
nebo více substitucemi). Z nich všechny izoláty jednotlivých nebo
dvojitých mutantů postrádající Q148H/K/R a celkem 10
z 24 izolátů s Q148H/K/R s dalšími substitucemi
souvisejícími s rezistencí vůči INSTI měly nejméně 2,5krát sníženou
citlivost na BIC; více než 2,5krát snížená citlivost na BIC byla
zjištěna u 14 z 24 izolátů, které obsahovaly substituce
G140A/C/S a Q148H/R/K v řetězci integrázy. Z těchto
14 izolátů jich mělo devět další mutace na L74M, T97A nebo E138A/K.
Kromě toho tzv. site‑directed mutanti s G118R
a T97A + G118R měli 3,4krát a 2,8krát sníženou citlivost
na BIC. Význam těchto dat o zkřížené rezistenci in
vitro je nutno potvrdit v klinické praxi. Biktegravir prokázal
ekvivalentní antivirovou aktivitu proti pěti klonům mutantů HIV‑1 rezistentních
vůči NNRTI, tři vůči NRTI a čtyři vůči PI v porovnání s kmenem
divokého typu [5].
U dosud neléčených pacientů (studie GS‑US‑380‑1489
a GS‑US‑380‑1490) a virologicky suprimovaných pacientů (studie GS‑US‑380‑1844
a GS‑US‑380‑1878) neměl žádný pacient užívající BIC HIV‑1 genotypovou nebo
fenotypovou rezistenci na BIC. Virologické selhání v průběhu léčby (n = 13, HIV‑1 RNA ≥ 200
kopií/μl) v týdnu 48 nebo při předčasném ukončení léčby hodnoceným lékem vzniklo z jiného důvodu. V době vstupu do studie mělo
šest dosud neléčených pacientů a jeden virologicky suprimovaný pacient
užívající BIC preexistující mutace související s rezistencí vůči INSTI
(šest subjektů s T97A a jeden dosud neléčený subjekt
s Q148H + G140S); všichni měli HIV‑1 RNA < 50
kopií/ml v týdnu 48 [3,7,8,9,10,11].
Závěr
Preklinická data prokázala srovnatelnou, v některých
testech i vyšší genetickou bariéru BIC s DTG oproti INSTI první
generace [2]. Biktegravir je dobře tolerován. Farmakokinetické interakce se
objevují zřídka[1]. Výsledky léčby BIC/F/TAF, DTG/ABC/3TC nebo DTG + F/TAF byly v týdnu 48 a 96
srovnatelné. Rychlý
pokles VL se ve čtvrtém týdnu blížil 80 %
[3,7,15,16]. Účinnost ani rychlost léčby nebyla ovlivněna subtypem HIV‑1.
Preexistující
lékové rezistence neovlivnily léčebné výsledky. Pacient s přenesenou
rezistencí k INSTI (G140S + Q148H),
fenotypově citlivý k BIC,
částečně k DTG,
byl léčen BIC/F/TAF. Hodnota
HIV‑1 RNA < 50
kopií/ml dosažená ve čtvrtém týdnu se udržela až do týdne 72 [13]. U žádného pacienta se nevytvořila
léčbu ohrožující rezistence ke studijním lékům BIC/F/TAF nebo k jiným
ART (NRTI, NNRTI)[3,7,15,16].
Kombinace
BIC/F/TAF se jeví jako účinná a spolehlivá s minimem nežádoucích
účinků. V průběhu roku 2019 lze očekávat zavedení tohoto přípravku mezi
stávající ART i v České republice. Kombinace BIC/F/TAF se
podává jednou denně s jídlem nebo bez jídla. Nedoporučuje se
u pacientů s hodnotou Clcr < 30 ml/min a u osob
s těžkou hepatopatií [1]. Před zahájením terapie se provede test
na virus hepatitidy B, stanoví se hodnota kreatininu, kreatininové
clearance, glykosurie, proteinurie. U osob s chronickou renální
insuficiencí se monitoruje rovněž sérová koncentrace fosfátů.
Článek vznikl za podpory společnosti Gilead Sciences
s.r.o.
Seznam použité literatury
- [1] SPC Biktarvy, 5/2019.
- [2] Tsiang M, Jones GS, Goldsmith J, et al. Antiviral Activity of Bictegravir (GS 9883), a Novel Potent HIV 1 Integrase Strand Transfer Inhibitor with an Improved Resistance Profile. Antimicrob Agents Chemother 2016; 60: 7086 7097.
- [3] Sax PE, Pozniak A, Montes ML, et al. Co formulated bictegravir, emtricitabine, and tenofovir alafenamide versus dolutegravir with emtricitabine and tenofovir alafenamide, for initial treatment of HIV 1 infection (GS US 380 1490): a randomised, double blind, multicentre, phase 3, non inferiority trial. Lancet 2017; 390: 2073‒2082.
- [4] Biktarvy US SmPC, 5/2019.
- [5] Deeks ED. Bictegravir/Emtricitabine/Tenofovir Alafenamide: A Review in HIV 1 Infection. Drugs 2018; 78: 1817–1828.
- [6] Spagnuolo V, Castagna A, Lazzarin A. Bictegravir. Curr Opin HIV AIDS 2018; 13: 326‒333.
- [7] Gallant J, Lazzarin A, Mills A, et al. Bictegravir, emtricitabine, and tenofovir alafenamide versus dolutegravir, abacavir, and lamivudine for initial treatment of HIV 1 infection (GS US 380 1489): a double blind, multicentre, phase 3, randomised controlled non inferiority trial. Lancet 2017; 390: 2063‒2072.
- [8] Wohl D, Yazdanpanah Y, Baumgarten A, et al. Phase III Randomized, Controlled Clinical Trial of Bictegravir in a Fixed Dose Combination, B/F/TAF, vs DTG/ABC/3TC in Treat-ment Naïve Adults at Week 96. IDWeek, Abstract 74246. October 3‒7, 2018, San Francisco, CA.
- [9] Stellbrink HJ, Arribas J, Stephens JL, et al. Phase III Randomized, Controlled Clinical Trial of Bictegravir Coformulated with FTC/TAF in a Fixed dose Combination (B/F/TAF) versus Dolutegravir (DTG) + F/TAF in Treatment naïve HIV 1 Positive Adults: Week 96. HIV Glasgow, Abstract 4185960. October 28‒31, 2018, Glasgow, UK.
- [10] Molina JM, Ward D, Brar I, et al. Switching to fixed dose bictegravir, emtricitabine, and tenofovir alafenamide from dolutegravir plus abacavir and lamivudine in virologically suppressed adults with HIV 1: 48week results of a randomised, double blind, multicentre, active controlled, phase 3, non inferiority trial. Lancet HIV 2018; http://dx.doi.org/10.1016/S2352 3018(18)30091 2
- [11] Daar ES, DeJesus E, Ruane P, et al. Efficacy and safety of switching to fixed dose bictegravir, emtricitabine, and tenofovir alafenamide from boosted protease inhibi-tor based regimens in virologically suppressed adults with HIV 1: 48week results of a randomised, open label, multicentre, phase 3, non inferiority trial. Lancet HIV 2018; http://dx.doi.org/10.1016/S2352 3018(18)30091 2
- [12] Gaur AH. Bictegravir/FTC/TAF Single Tablet Regimen in Adolescents & Children: Week 48 results. CROI 2019. Abstract 46.
- [13] HIV Drug Interactions. University of Liverpool. Dostupné na: https://www.hiv druginteractions.org/checker
- [14] Podzamczer D, Stellbrink A, Orkin C, et al. HIV Drug Therapy 2018. Glasgow, UK. Poster 119.
- [15] Wohl DA, Yazdanpanah Y, Baumgarten A, et al. Bictegravir combined with emtricitabine and tenofovir alafenamide versus dolutegravir, abacavir, and lamivudine for initial treatment of HIV 1 infection: week 96 results from a randomised, double blind, multicentre, phase 3, non inferiority trial. Lancet HIV 2019; http://dx.doi.org/10.1016/S2352 3018(19)30077 3.
- [16] Stellbrink HJ, Arribas JR, Stephens JL, et al. Co formulated bictegravir, emtricitabine, and tenofovir alafenamide versus dolutegravir with emtricitabine and tenofovir ala-fenamide for initial treatment of HIV 1 infection: week 96 results from a randomised, double blind, multicentre, phase 3, non inferiority trial. Lancet HIV 2019; http://dx.doi.org/10.1016/S2352 3018(19)30080 3.
- [17] White K, Niedziela Majka A, Novikov N, et al. BIC Dissociation from G140S Q148H Integrase. CROI 2017, Poster 497.
- [18] SPC Isentress, 5/2019.
- [19] SPC Tivicay, 5/2019.