Biosimilars – podobné biologické léčivé přípravky
Po skončení patentové ochrany originálních biologických léčivých přípravků se na farmaceutickém trhu objevují tzv. biosimilars. Biosimilars jsou biologické léčivé přípravky podobné původním, originálním přípravkům. Jako všechny biologické přípravky, které jsou produktem živých organismů, jsou přirozeně proměnlivé. Biosimilars není vhodné označovat termínem biogenerikum vzhledem k obecné proměnlivosti těchto přípravků a ke skutečnosti, že je nemožné přesně definovat jejich strukturu. Na rozdíl od klasických generických přípravků se pro posouzení účinnosti a bezpečnosti biosimilars vyžaduje v rámci registračního řízení doložení preklinických a klinických studií porovnávajících biosimilars s originálním přípravkem se zvláštním zřetelem na charakterizaci imunogenního potenciálu produktu.
Biologické léčivé přípravky
Biologická léčiva jsou taková léčiva, při jejichž přípravě se používají živé organismy nebo jejich produkty a jejichž strukturu nelze zcela přesně definovat. Jsou to obvykle proteiny nebo polypeptidy. Jejich různorodost je dána prostorovým tvarem molekuly a typem a délkou každé navázané cukerné nebo uhlovodíkové skupiny. Je výsledkem přirozených procesů v hostitelské buňce, které modifikují proteiny během jejich produkce, aby byly chráněny při transportu přes jednotlivé buněčné prostory. K modifikaci proteinů dochází také v průběhu jejich extrakce, čištění, formulace lékové formy a skladování. Vedle této heterogenity související s vlastním produktem vznikají z hostitelských buněk a kultivačních médií během výrobního procesu nečistoty ovlivňující vlastnosti biologického přípravku [1–5].
Změny v navázaných cukerných jednotkách mohou vést k velkému rozdílu v účinnosti, imunogenitě i biologickém poločasu léčivé látky. Např. rozdílná glykosylace proteinů epoetinu alfa a epoetinu beta je minimálně částečně zodpovědná za pozorované rozdíly v biologické aktivitě těchto dvou molekul [6].
Biologická léčiva reprezentují jedno z nejrychleji rostoucích odvětví farmaceutického průmyslu s ročním přírůstkem 12–15 % [3]. Nejvíce se rozvíjející oblast moderních biologických léčiv představují monoklonální protilátky, jejichž hlavní terapeutické využití spočívá v léčbě chorob spojených s poruchou imunitního systému, především rakovinných a zánětlivých onemocnění. Monoklonální protilátky jsou buď chimérické, tzn. že jsou tvořeny zčásti živočišným proteinem (mají v názvu příponu „-ximab“), humanizované, v nichž je přítomen převážně lidský protein (s příponou „-zumab“), humánní, které jsou tvořeny pouze lidským proteinem (přípona „-umab) případně dnes již méně používané myší protilátky (s příponou „-omab“) [2, 7].
K přípravě většiny substancí biologických léčiv je použito rekombinantní DNA (rDNA) technologie. Obecně se dají rozdělit do tří skupin:
- Biologické produkty obsahující biotechnologicky získané proteiny jako účinné látky.
- Imunologické preparáty, jako jsou vakcíny a alergeny.
- Produkty získané z krve a plazmy a jejich rekombinantní alternativy [1–3].
Výhody a nevýhody moderních biologických léčiv
Nespornými výhodami monoklonálních protilátek a dalších proteinů připravených rDNA technologií jsou:
- jejich specifický způsob účinku, který výrazně snižuje riziko vedlejších nežádoucích účinků;
- lépe zasahují do podstaty patologického procesu, často působí přímo na příslušné receptorové místo, transportní protein nebo enzym. Klasická chemická léčiva většinou nejsou schopna tak cíleného působení, např. většina cytostatik ovlivní všechny buňky schopné mitózy;
- vývoj biologického léčiva od zahájení preklinických studií po přípravu registrační dokumentace je kratší ve srovnání s chemickým léčivem.
- Na druhé straně je použití biologických léčiv spojeno s některými vážnými problémy:
- vysoká cena léčby těmito přípravky, která v případě monoklonálních protilátek řádově převyšuje cenu obdobné léčby chemickými léčivy. Důsledkem jsou významně nižší (o dva až tři řády) počty pacientů léčených těmito přípravky;
- velkoobjemová výroba spojená s vyššími výrobními náklady, a to jak surovinovými, tak personálními. Rovněž v délce výrobního cyklu jsou značné rozdíly – u biotechnologických léčiv se pohybuje v rozmezí 25–50 týdnů, zatímco v případě chemických léčiv trvá výrobní cyklus v průměru 3 týdny;
- příliš blízký vztah k přirozeným ligandům může způsobit nepřiměřenou odpověď imunitního systému. Po terapii těmito léčivy se častěji vyskytují autoimunitní onemocnění. Tyto problémy jsou sice mírnější u humanizovaných protilátek, ale ani ony tento problém zcela neřeší. Objevují se rovněž nežádoucí efekty, které nesouvisí přímo s mechanismem účinku biologického léčiva, např. progresivní multifokální encefalopatie po rituximabu [2, 8].
Podobné biologické léčivé přípravky (biosimilars)
Existence podobných biologických léčivých přípravků snižuje cenu originálních biologických přípravků o 20 %. Uvedení biosimilars na farmaceutický trh v Evropské unii je možné na základě úspěšného absolvování tzv. centralizované procedury (je povinná pro registraci léčivých přípravků vyrobených špičkovou technologií, zvláště pro přípravky, které jsou výsledkem biotechnologických procesů) a následného schválení Evropskou komisí na základě doporučení Evropské lékové agentury [9]. Na rozdíl od klasických generických přípravků jsou u biosimilars vyžadovány srovnávací studie prokazující kvalitu, bezpečnost a účinnost podobnou té, jakou má originální referenční léčivý přípravek [1]. Doložení účinnosti a bezpečnosti je jedinečné pro každý podobný biologický léčivý přípravek a pro každou účinnou látku, kterou obsahuje, s ohledem na použité metody během vývoje přípravku a klinického hodnocení a je zakotveno v evropských směrnicích [10, 11]. Za klíčové z hlediska provedení dalších studií je považováno posouzení, zda byl podobný biologický přípravek během svého vývoje hodnocen ve stejné lékové formě, síle a zda se použila stejná cesta podání jako u referenčního přípravku a používal-li se ve všech studiích stejný referenční přípravek registrovaný v Evropské unii [12]. Např. současné evropské směrnice vyžadují u podobných léčivých přípravků s růstovým hormonem šesti- až dvanáctiměsíční srovnávací studii porovnávající účinnost a bezpečnost v rámci klinického hodnocení fáze III [13, 14]. Standardní postup pro generická léčiva s definovanou strukturou, tj. identifikace aktivní substance a bioekvivalence lékové formy, je pro ně nepoužitelný [15]. Je požadován mnohem obsáhlejší a finančně náročnější vývoj s použitím nejmodernějších analytických metod a rozsáhlejší registrační dokumentace. Z tohoto důvodu se považuje za matoucí termín „biogenerikum“, jako vhodnější se doporučují termíny „podobný biologický léčivý přípravek“ nebo „biosimilars“. Jedním z klíčových aspektů pro úspěšné schválení podobného biologického léčivého přípravku je jeho rozsah proměnlivosti v účinnosti a čistotě, který by se měl pohybovat v mezích originálního přípravku. Cena vývoje biosimilars je minimálně desetkrát vyšší oproti ceně vývoje klasického generického léčiva. Po schválení podobného biologického přípravku je vyžadována specifická farmakovigilance v postregistračním období. Souhrn údajů o přípravku, označení na obalu, příbalová informace a informace o výrobci biologické léčivé látky, o výrobci, který odpovídá za propouštění šarží, a o podmínkách registrace schváleného biologického přípravku jsou volně přístupné na stránkách Evropské lékové agentury jako tzv. EPARs – European Public Assessment Reports [3, 4].
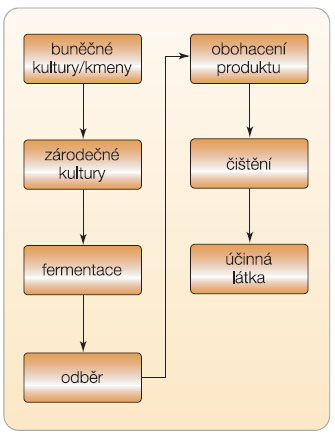 Příprava biosimilars začíná s naklonovanými buněčnými liniemi (mají vložen cílový gen v buňce) nebo se zárodečnou kulturou připravenou k fermentaci (obr. 1). Následuje fermentace, odběr vzorku, obohacení produktu, čištění a izolace účinné látky. Buněčná linie nebo kultura může být vyvinuta výrobcem, zakoupena z jiné společnosti nebo vyvinuta ve spolupráci s jinou společností. Tímto způsobem je možné zkrátit vývoj biosimilars až o 2 roky [3].
Příprava biosimilars začíná s naklonovanými buněčnými liniemi (mají vložen cílový gen v buňce) nebo se zárodečnou kulturou připravenou k fermentaci (obr. 1). Následuje fermentace, odběr vzorku, obohacení produktu, čištění a izolace účinné látky. Buněčná linie nebo kultura může být vyvinuta výrobcem, zakoupena z jiné společnosti nebo vyvinuta ve spolupráci s jinou společností. Tímto způsobem je možné zkrátit vývoj biosimilars až o 2 roky [3].
Příprava podobného biologického léčivého přípravku je doprovázena řadou problémů, jež jsou spojeny se složitou strukturou biotechnologických léčiv, kterou nelze zcela přesně definovat, jako je tomu u chemických léčiv. Mezi odborníky zatím převládá názor, že vzhledem ke složitosti prostorové konfigurace molekul o velikosti 103 až 106 D nebude možno dosáhnout stejné struktury generické a originální substance. Všechny biologické přípravky jsou přirozeně proměnlivé, protože jsou produktem živých organismů. Tato variabilita ve vlastnostech určitého biologického přípravku existuje nejen mezi různými výrobci daného přípravku, ale objevuje se také se změnou technologie a dokonce i mezi jednotlivými výrobními šaržemi. V důsledku je nutné srovnání nejen v kvalitě, ale i v bezpečnosti a účinnosti s autorizovaným referenčním standardem se zvláštním zřetelem na charakterizaci imunogenního potenciálu produktu. V mnoha případech je u biosimilars vyžadováno klinické hodnocení na několika stech pacientů [2, 3].
Imunogenita
Imunogenita je schopnost specifické látky navodit produkci protilátek lidského těla. Kromě kolonie stimulujícího faktoru pro granulocyty prokazují na rozdíl od konvenčního přípravku všechny biologické léčivé přípravky větší schopnost vyvolat imunitní reakce indukcí tvorby protilátek. Imunogenita léčivých proteinů je založena na dvou rozdílných mechanismech. K první, klasické reakci na cizí proteiny dochází po podání biologických léčiv bakteriálního nebo rostlinného původu, jako je streptokináza nebo aspargináza. Druhý mechanismus, kterým dochází k indukci protilátek, je založen na prolomení imunitní tolerance k antigenům tělu vlastním. To je mechanismus, který nasměruje protilátky proti homologům lidských proteinů, jako jsou interferony, interleukin-2, epoetin. Proč a jakým způsobem dochází k prolomení imunitní tolerance, není dosud zcela vysvětleno. Začíná být jasné, že je to právě agregace, která je u léčiv na bázi proteinů nejdůležitějším faktorem vedoucím k prolomení tolerance B buněk, ačkoli další faktory mohou k tomuto jevu přispívat. Dokonce i malé množství agregátů může být postačující k vyvolání imunitní odpovědi. Agregací se buď odhalují nové epitopy (antigenní determinanty, konkrétní oblasti antigenu, na niž se vážou protilátky), které jsou rozpoznány jako cizí, nebo vzniká nové rozmístění známých epitopů, jež vede k prolomení tolerance. Velmi silnou schopnost indukovat protilátkovou odpověď i při absenci T buněk mají vysoce organizované proteinové struktury (např. nedenaturované agregáty bílkovin). Naopak cukerné nebo polyethylenglykolové jednotky navázané na proteinovém řetězci mohou vést ke snížení imunogenity léčivého přípravku. Velmi málo je dosud známo o vztazích mezi chemickými nebo konformačními změnami ve struktuře proteinů a imunogenitou [4, 16–18].
U mnoha pacientů není podání biologického přípravku doprovázeno nežádoucí imunitní reakcí. Nicméně, existuje potenciál pro celkové závažné nežádoucí imunitní reakce, jako je alergie a anafylaxe. Navíc mohou biologické léčivé přípravky vyvolat reakce, které vedou ke ztrátě léčebného účinku s vážnými terapeutickými následky (tvorba protilátek je často doprovázena terapeutickým selháním). Imunogenní potenciál je ovlivněn jak výrobním procesem a formulací vlastního biologického přípravku (např. změnou pomocných látek nebo postupu výroby lze zabránit vzniku agregátů léčivé látky), tak i faktory vztaženými na konkrétního pacienta, nemoc a léčbu, jako jsou např. cesta podání nebo snížená imunitní odpověď u pacientů léčených cytostatiky. Jiným příkladem mohou být výsledky klinických studií, v nichž byl aplikován infliximab jednorázově nebo opakovaně v dávkovém rozmezí 1 až 20 mg/kg a ve kterých byly zjištěny protilátky na infliximab u 14 % pacientů léčených různými imunosupresivy a u 24 % pacientů bez imunosupresivní léčby [8]. Uvedené faktory jsou pečlivě hodnoceny během vývoje a posuzovány u všech biologických přípravků, včetně biosimilars. Přesné postupy hodnocení imunogenního potenciálu pro daný biologický přípravek jsou zakotveny v evropských směrnicích [3].
Závěr
Snížení výdajů ve zdravotnictví je žhavým politickým tématem v mnoha zemích. Jednou z možností, jak dosáhnout tohoto cíle, je podpora užívání generických léčivých přípravků. U biosimilars je však třeba mít na paměti, že se jedná o biologické léčivé přípravky charakterizované svým vlastním kvalitativním profilem a dlouhodobé důsledky těchto rozdílů vzhledem k originálnímu přípravku nejsou detailně prozkoumány. Podrobnější informace nám jistě přinesou dlouhodobá postmarketingová sledování ve všech relevantních populacích. Vzhledem k tomu, že se výskyt imunitních reakcí po podání vzájemně podobných biologických léčivých přípravků může lišit, není vhodné jejich nepromyšlené zaměňování, ať už jde o záměnu biosimilars za originální přípravky, nebo naopak.
Seznam použité literatury
- [1] CHMP/437/04. EMEA, Committee for medicinal products for human use: Guideline on similar biological medicinal products.
- [2] Kuchař M. Výzkum a vývoj léčiv. 1. vyd. VŠCHT Praha, 2008: 153–158.
- [3] European Generic Medicines Association. EGA Handbook on biosimilar medicines. European Generic Medicines Association Brussels, 2008. 24 s.
- [4] Schellekens H. The First Biosimilar Epoetin: But How Similar Is It? Clin J Am Soc Nephrol 2008; 3: 174–178.
- [5] Tucker J, Yakatan S, Yakatan S. Biogenerics 2007: How far have we come? J Commer Biotechnol 2008; 14: 56–64.
- [6] Storring PL, Tiplady RJ, Gaines Das RE, et al. Epoetin alfa and beta differ in their erythropoietin isoform compositions and biological properties. Br J Haematol 1998; 100: 79–89.
- [7] Marek J, et al. Farmakoterapie vnitřních nemocí. 3. vyd. Praha, Grada, 2005. 773 s.
- [8] Mikroverze AISLP – ČR verze 2010.1 – 1. 1. 2010.
- [9] Regulation (EC) No 726/2004 of the European Parliament and of the Council of 31 March 2004 laying down Community procedures for the authorisation and supervision of medicinal products for human and veterinary use and establishing a European Medicines Agency.
- [10] http: //www.ema.europa.eu/htms/human/human-guidelines/multidiscipline.htm, staženo 4. 3. 2010.
- [11] Pavlovic M, Girardin E, Kapetanovic L, et al. Similar biological medicinal products containing recombinant human growth hormone: European regulation. Horm Res 2008; 69: 14–21.
- [12] Ronco C. Is the advent of biosimilars affecting the practice of nephrology and the safety of patients? In: Ronco C, Cruz DN (eds). Hemodialysis – from basic research to clinical trials. Contributions to Nephrology. Basel: Karger 2008; 161: 261–270.
- [13] Saenger P. Current status of biosimilar growth hormone. Int J Pediatr Endocrinol 2009; 2009: 370329.
- [14] CHMP/94528/05. EMEA, Committee for medicinal products for human use: Annex to guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: Non-clinical and clinical issues – Guidance on similar medicinal products containing somatropin.
- [15] Vetchý D. Klinické a ekonomické dopady generické substituce. Practicus 2008; 10: 27–30.
- [16] Rosenberg AS. Effects of protein aggregates: An immunologic perspective. Aaps J 2006; 8: E501–E507.
- [17] Hermeling S, Crommelin DJ, Schellekens H, Jiskoot W. Structure-immunogenicity relationships of therapeutic proteins. Pharm Res 2004; 21: 897–903.
- [18] Schellekens H. How similar do 'biosimilars' need to be? Nat Biotechnol 2004; 22: 1357–1359.