Enzymová substituční terapie pacientů s mukopolysacharidózami
Souhrn:
Mukopolysacharidózy (MPS) jsou lyzosomální střádavá onemocnění způsobená nedostatečnou aktivitou některého z lyzosomálních hydrolytických enzymů podílejících se na postupné degradaci glykosaminoglykanů (původně mukopolysacharidů). Mají řadu společných rysů, jednotlivé typy se však vzájemně odlišují. Klinické projevy onemocnění se vyvíjejí postupně a jsou závislé na typu MPS i na její tíži. Patří mezi ně hepatomegalie/splenomegalie, pupeční a/nebo tříselná kýla, kraniofaciální dysmorfie ve smyslu hrubých rysů obličeje, opakované infekty horních cest dýchacích, záněty středouší, poruchy sluchu, chlopenní vady či kombinované plicní postižení. Konstantní je kostní postižení s poruchou růstu, s deformitami páteře a s charakteristickým rentgenovým nálezem dysostosis multiplex. Variabilní je kognitivní postižení. Výraznou změnu v prognóze pacientů s MPS přineslo zavedení enzymové substituční terapie ((enzyme replacement therapy, ERT). Tato léčba zmírňuje viscerální postižení a zlepšuje kvalitu života pacientů, neovlivňuje však postižení CNS. Pro maximální účinek enzymové substituční terapie je důležitá časná diagnostika a časné zahájení léčby.
Key words: mucopolysaccharidoses – enzyme replacement therapy.
Summary:
Mucopolysaccharidoses (MPS) are lysosomal storage disorders caused by low activity of some of lysosomal hydrolytic enzymes degrading the glycosaminoglycans (originally mucopolysaccharides). All forms of the disease share a lot of common features, although particular MPS types differ one from each other. Clinical signs evolve gradually and depend on the MPS type and its severity. They include hepato-/splenomegaly, umbilical and/or inguinal hernia, craniofacial dysmorphy with coarse facial features, recurrent upper respiratory tract infections, middle ear infections, hearing disorder, cardiac valve involvement or combined pulmonary involvement. Bone involvement with short stature, backbone deformities and characteristic X-ray changes outlined as dysostosis multiplex are constant features. Cognitive impairment is variable. The enzyme replacement therapy (ERT) has brought a significant change to the prognosis of patients with MPS. It improves the visceral involvement together with the patients‘ quality of life. It has no effect upon CNS impairment. The early diagnosis and therapy introduction is essential for its maximal benefit.
Úvod
Mukopolysacharidózy (MPS) jsou dědičné metabolické poruchy patřící do skupiny více než sedmdesáti známých lyzosomálních střádavých onemocnění [1]. Lidstvo provázejí od nepaměti. Vyobrazení pacientů s výrazem nápadně připomínajícím MPS lze vysledovat do středověku. Kanadskou rodinu chlapce s MPS typu II popsal ve svém románu severoamerický spisovatel Washington Irving (1783–1859). To, že se jednalo právě o tuto nemoc, lze doložit i na základě klasického rodokmenu postižených členů rodiny. První doložený klinický popis MPS pochází až z pera skotského doktora Charlesa Huntera z roku 1917. Ve své nové kanadské vlasti publikoval kasuistiky dvou bratrů s hrubými rysy obličeje, zvětšeným břichem a s kostní dysplazií. Hunterův syndrom (MPS II) se tak stal jedním z jedenácti dnes známých typů a podtypů MPS [2].
Primárním patofyziologickým podkladem MPS je střádání kyselých mukopolysacharidů, nověji glykosaminoglykanů (GAG), v tkáních organismu. Příčinou je nedostatečná aktivita některého z lyzosomálních hydrolytických enzymů podílejících se na postupné degradaci GAG. Kromě samotného střádání GAG nejenom v lyzosomech, ale i v dalších buněčných kompartmentech a v extracelulární matrix hrají zásadní roli indukované sekundární patofyziologické kaskády. Ty zahrnují aberantní genovou expresi, indukci prozánětlivého stavu, oxidačního stresu, aberantní autofagocytózu s nahromaděním poškozených mitochondrií a s indukcí buněčné smrti. Nahromaděné makromolekuly aktivují či inhibují přenos signálu, mění lokalizaci receptorů a modifikují jejich odpověď. Defektní je homeostáza vápníkových iontů, syntetická funkce buňky, maturace proteinů a buněčný transport [3]. Mukopolysacharidózy jsou děděny autosomálně recesivně s výjimkou MPS typu II, která je vázána na chromosom X. Souhrnná incidence mukopolysacharidóz v České republice (ČR) je vyjádřena poměrem 1 : 26 882 narozených dětí [4].
Klinická manifestace
Mukopolysacharidózy jsou onemocnění progresivní a multisystémová s vysokou klinickou variabilitou. Mají řadu společných rysů, jednotlivé typy se však od sebe vzájemně liší (tab. 1) [5–7]. Časně se většinou manifestují těžké formy s nepříznivou prognózou, později pak většinou mírné formy s přežíváním do dospělosti [8]. Klinické projevy onemocnění se vyvíjejí postupně a jsou závislé na typu onemocnění i na jeho tíži. Z porodnice odchází většina dětí jako zdraví novorozenci. První příznaky se mohou objevit často již v kojeneckém nebo v batolecím období, jsou vesměs nespecifické a k diagnóze může vést právě jejich kombinace. Jedná se 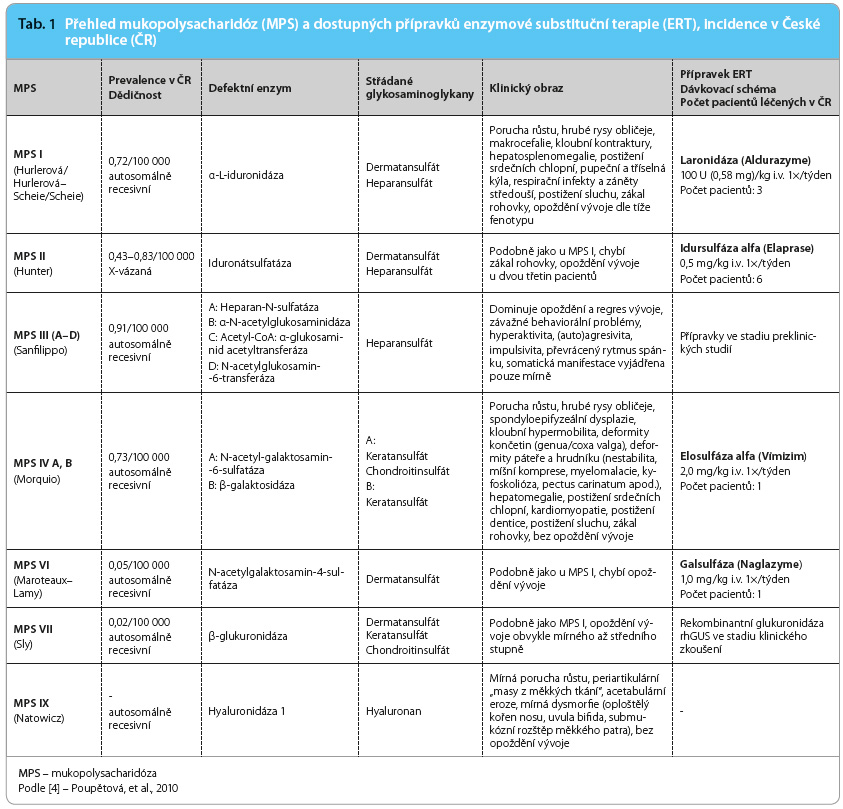 zejména o pupeční nebo tříselnou kýlu, větší obvod hlavy, opakované záněty horních cest dýchacích či středouší, o chronické vodnaté průjmy a poruchu sluchu. U rozvinutého onemocnění pak bývají patrné hrubší rysy obličeje s klenutým čelem, prominujícími nadočnicovými oblouky, širokým kořenem nosu, antevertrovanými nostrilami a s plnými rty (obr. 1 2 3). Zejména u některých mírných forem a u MPS typu III mohou být dysmorfické rysy pouze stěží rozpoznatelné. Kratší nosní průchody a makroglossie souvisejí s chronickou rýmou, s hlasitým dýcháním, s ronchopatií, s hypertrofií nosní i patrových mandlí a s opakovanými záněty středouší. Mohou být přítomny tvarové změny zubů. Hlas může být nápadně hluboký a chraplavý. Přítomna je hepatomegalie a splenomegalie, a to zejména u MPS typu I, II a VI. Střádání GAG probíhá i na srdečních chlopních s rozvojem chlopenních vad, sekundárně se může rozvinout
zejména o pupeční nebo tříselnou kýlu, větší obvod hlavy, opakované záněty horních cest dýchacích či středouší, o chronické vodnaté průjmy a poruchu sluchu. U rozvinutého onemocnění pak bývají patrné hrubší rysy obličeje s klenutým čelem, prominujícími nadočnicovými oblouky, širokým kořenem nosu, antevertrovanými nostrilami a s plnými rty (obr. 1 2 3). Zejména u některých mírných forem a u MPS typu III mohou být dysmorfické rysy pouze stěží rozpoznatelné. Kratší nosní průchody a makroglossie souvisejí s chronickou rýmou, s hlasitým dýcháním, s ronchopatií, s hypertrofií nosní i patrových mandlí a s opakovanými záněty středouší. Mohou být přítomny tvarové změny zubů. Hlas může být nápadně hluboký a chraplavý. Přítomna je hepatomegalie a splenomegalie, a to zejména u MPS typu I, II a VI. Střádání GAG probíhá i na srdečních chlopních s rozvojem chlopenních vad, sekundárně se může rozvinout 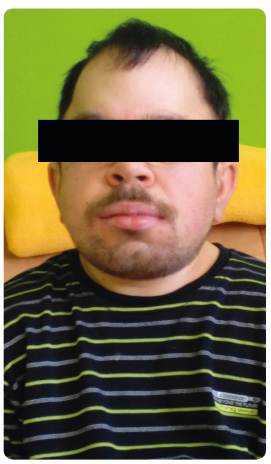 kardiomyopatie. Plicní postižení je kombinované s obstrukční i s restrikční poruchou. Kůže může být zhrubělá, často s hypertrichózou, s hustým obočím, s nižší vlasovou hranicí, u MPS typu II je popisován specifický obraz „pomerančové kůže“ lokalizované zejména na zádech. Prakticky konstatní je postižení skeletu s poruchou růstu, s deformitami páteře, s d
kardiomyopatie. Plicní postižení je kombinované s obstrukční i s restrikční poruchou. Kůže může být zhrubělá, často s hypertrichózou, s hustým obočím, s nižší vlasovou hranicí, u MPS typu II je popisován specifický obraz „pomerančové kůže“ lokalizované zejména na zádech. Prakticky konstatní je postižení skeletu s poruchou růstu, s deformitami páteře, s d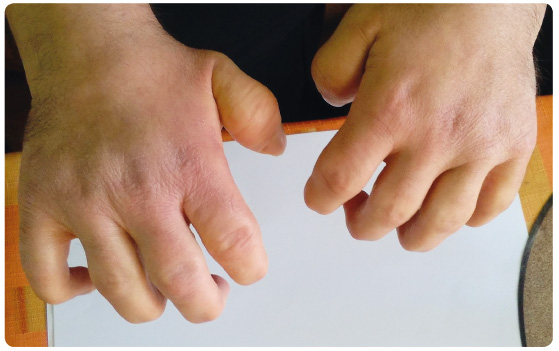 ysplazií kyčelních a kolenních kloubů [6,7]. Odpovídající rentgenový nález se označuje termínem dysostosis multiplex. Sella turcica na bočním rentgenovém snímku lebky je deformovaná a protažená do tvaru písmene J. Dlouhé kosti jsou zkrácené s postižením metafýz i epifýz. Změny na kostech ulny a radia vedou k deformitě ve tvaru písmene V. Těla obratlů mají typický ovoidní tvar, žebra se ventrálně rozšiřují. Pánev je hypoplastická a hlavice i krčky stehenní kosti jsou deformované. Dominantní postižení skeletu s těžkou poruchou růstu je typické pro MPS typu IV. Charakteristická pro MPS bývá kloubní ztuhlost s vývojem kontraktur velkých i malých kloubů a s následným omezením hrubé i jemné motoriky, často s rozvojem drápovité ruky. Výjimkou je opět MPS typu IV, u které se
ysplazií kyčelních a kolenních kloubů [6,7]. Odpovídající rentgenový nález se označuje termínem dysostosis multiplex. Sella turcica na bočním rentgenovém snímku lebky je deformovaná a protažená do tvaru písmene J. Dlouhé kosti jsou zkrácené s postižením metafýz i epifýz. Změny na kostech ulny a radia vedou k deformitě ve tvaru písmene V. Těla obratlů mají typický ovoidní tvar, žebra se ventrálně rozšiřují. Pánev je hypoplastická a hlavice i krčky stehenní kosti jsou deformované. Dominantní postižení skeletu s těžkou poruchou růstu je typické pro MPS typu IV. Charakteristická pro MPS bývá kloubní ztuhlost s vývojem kontraktur velkých i malých kloubů a s následným omezením hrubé i jemné motoriky, často s rozvojem drápovité ruky. Výjimkou je opět MPS typu IV, u které se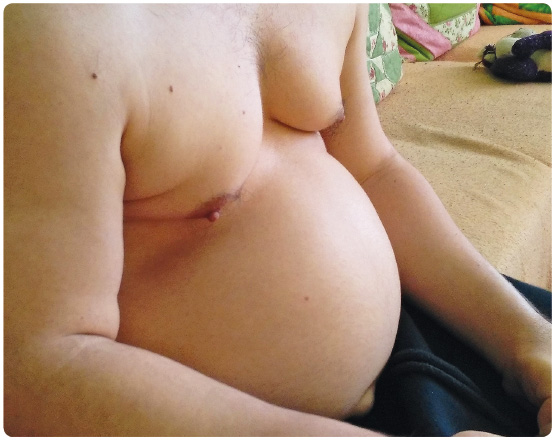 naopak vyskytuje spíše hypermobilita a kloubní laxita. U všech typů MPS dochází k rizikovému střádání v oblasti krční páteře s nebezpečím rozvoje cervikální myelopatie a atlantookcipitální instability s přísnou kontraindikací výrazného záklonu hlavy s rizikem náhlé smrti zejména při intubaci [9]. Opoždění a často i regres vývoje jsou typické pro těžké formy MPS typu I, II a III. Nevyskytuje se naopak u MPS typu IV a VI. Pro MPS typu II a III je typická psychiatrická symptomatologie s poruchami chování (hyperaktivita, agresivita, afektivní záchvaty, střídání emocí). Rodinu dítěte s MPS typu III pak vyčerpává absence cirkadiánní rytmicity s převráceným spánkovým režimem [10]. Děti s těžkými formami MPS typu I, II a VI mohou rozvinout hydrocefalus a epilepsii. Častým problémem je syndrom karpálního tunelu.
naopak vyskytuje spíše hypermobilita a kloubní laxita. U všech typů MPS dochází k rizikovému střádání v oblasti krční páteře s nebezpečím rozvoje cervikální myelopatie a atlantookcipitální instability s přísnou kontraindikací výrazného záklonu hlavy s rizikem náhlé smrti zejména při intubaci [9]. Opoždění a často i regres vývoje jsou typické pro těžké formy MPS typu I, II a III. Nevyskytuje se naopak u MPS typu IV a VI. Pro MPS typu II a III je typická psychiatrická symptomatologie s poruchami chování (hyperaktivita, agresivita, afektivní záchvaty, střídání emocí). Rodinu dítěte s MPS typu III pak vyčerpává absence cirkadiánní rytmicity s převráceným spánkovým režimem [10]. Děti s těžkými formami MPS typu I, II a VI mohou rozvinout hydrocefalus a epilepsii. Častým problémem je syndrom karpálního tunelu.
Diagnostika mukopolysacharidóz
Diagnostika pacienta s podezřením na MPS je náročná a pro možnou podobnost s jinými dědičnými metabolickými poruchami by měla probíhat na pracovišti se zkušeností v dané oblasti. Klinické podezření je nutno potvrdit na biochemické a molekulárně genetické úrovni. Prvním krokem je kvantitativní stanovení vylučování GAG v moči. Zejména u lehkých forem však může být výsledek negativní nebo hraniční a vyšetření je nutno opakovat z 24hodinového sběru. V případě průkazu zvýšené exkrece GAG v moči se provádí elektroforetické vyšetření s rozdělením mukopolysacharidů na jednotlivé frakce. Zvýšené vylučování dermatansulfátu a heparansulfátu nacházíme u MPS typu I, II a VI, zvýšené vylučování heparansulfátu u MPS typu III a keratansulfátu u MPS typu IV [11]. K potvrzení diagnózy jednotlivých typů MPS na biochemické úrovni je nutné stanovit aktivitu jednotlivých enzymů v izolovaných leukocytech, v buněčné kultuře kožních fibroblastů nebo nově v suché krevní kapce. Po stanovení diagnózy je potřeba vypracovat detailní rodokmen, identifikovat členy rodiny s možným rizikem přenašečství a nabídnout dostupné genetické poradenství [11]. Vzhledem k závažnosti onemocnění je zcela zásadní empatický a citlivý přístup při sdělování diagnózy s nutností dalšího vedení rodiny.
Enzymová substituční terapie
Výraznou změnu pro prognózu dětí s MPS přineslo zavedení enzymové substituční terapie (enzyme replacement therapy, ERT). První myšlenky náhrady deficitního enzymu funkčním proteinem se objevily záhy po pochopení podstaty střádavých onemocnění v šedesátých letech minulého století [12,13]. Klíčovým pro další vývoj byl náhodný objev tzv. cross‑correction fenoménu. V americké laboratoři profesorky Elizabeth F. Neufeldové došlo nedopatřením ke smíchání buněčných kultur fibroblastů pacientů s Hunterovým a Hurlerové syndromem (MPS typu I a II). Známky lyzosomálního střádání smíšené linie během několika hodin vymizely [14]. Byla vyslovena hypotéza vzájemné výměny chybějícího genového produktu označovaného jako „corrective factor“. Jedná se o malé množství enzymu, který nebyl v buňce správně směrován do lyzosomu a je buňkou vylučován do extracelulární matrix. Pro korekci defektního metabolismu překvapivě stačí velmi malé množství enzymu představující v některých případech pouze 1–5 % normální enzymové aktivity [15].
Proteiny určené pro lyzosom jsou označeny specifickým signálem – molekulou manóza‑6‑fosfátu. Receptory pro tyto specifické signály existují nejen na povrchu lyzosomů, ale i na plazmatické membráně buněk, což umožňuje vychytávání daného enzymu buňkou a jeho následný transport do lyzosomu [16]. Schopnost buněk internalizovat enzym z extracelulárního prostředí a využívat jej je pro fungování ERT nezbytná. Objasnění významu manóza‑6‑fosfátových receptorů proto bylo dalším důležitým krokem umožňujícím uvedení ERT do praxe.
Pokusy s ERT se datují do počátku sedmdesátých let 20. století. Enzym byl postupně izolován z plazmy, z králičího mléka, moči či z kultur Aspergillus niger. Prvním schváleným přípravkem se stala glukocerebrosidáza pro léčbu pacientů s Gaucherovou nemocí v roce 1991. Byla získávána purifikací z lidských placent a následnou glykosylací pro expozici manózového zbytku [17]. Enzymový výtěžek z placent však byl nedostatečný, navíc s rizikem infekčních komplikací. Řešení přinesla produkce rekombinantního enzymu v buněčné linii ovarií čínských křečků (Chinese Hamster Ovary cells, CHO). Jednalo se opět o glukocerebrosidázu ošetřenou exoglykosidázami [18]. Linie CHO buněk umožnila produkci enzymů v potřebné kvantitě i kvalitě i pro řadu dalších onemocnění: α‑galaktosidázy pro Fabryho nemoc [19], α-L‑iduronidázy pro MPS typu I [20] a α‑glukosidázy pro Pompeho nemoc [21]. Dnes jsou rekombinantní enzymy produkovány nejen linií CHO buněk, ale rovněž v buněčných kulturách lidských fibroblastů, na bakteriálních platformách či nověji v modifikovaných rostlinných buněčných liniích [22].
Enzymová substituční terapie je v současné době dostupná pro čtyři typy MPS – I, II, IV A a VI (tab. 1). V případě těžké formy MPS typu I, nemoci Hurlerové, je při ještě zachovalém vývoji indikována transplantace hematopoetických kmenových buněk (hematopoietic stem cell transplantation, HSCT) v kombinaci s ERT s nadějí na stabilizaci kognitivního postižení [6]. Výsledky HSCT u pacientů s MPS typu II nebyly příznivé. Dětští i dospělí pacienti s MPS, jimž je podávána ERT, jsou sledováni a léčeni ve specializovaném centru na Klinice dětského a dorostového lékařství 1. LF UK a VFN v Praze. Léčba probíhá ve spolupráci s dětskými odděleními v místech bydliště pacientů. Ampule enzymu se ředí do 100 ml fyziologického roztoku podávaného v nitrožilní infuzi trvající 2–3 hodiny. Iniciální nižší rychlost infuze se postupně zvyšuje podle připraveného protokolu. Zejména u prvních aplikací a při pozitivní anamnéze alergické reakce je vhodné pacienta premedikovat antihistaminiky v odpovídající dávce. Pacient je během aplikace monitorován. Prvních deset aplikací ERT se u dětí pro zvýšené riziko 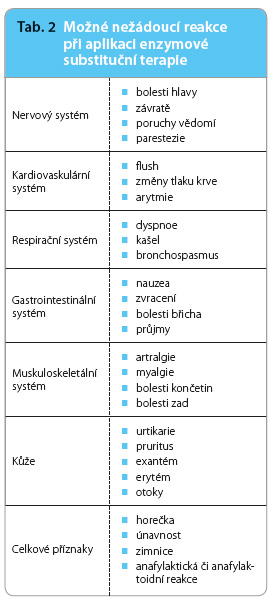 nežádoucích, zejména alergických reakcí (tab. 2) podává během hospitalizace. Infekt doprovázený horečkou nebo afekce, která může imitovat nežádoucí účinek, např. nově vzniklá vyrážka, je kontraindikací podání přípravku. Na možnost nežádoucích účinků je však nutno být připraven po celou dobu podávání léčby. V takovém případě je nezbytné zastavit aplikaci, zhodnotit závažnost reakce, podat infuzi krystaloidů, dle tíže reakce podat antihistaminika, antipyretika, ev. kortikoidy a adrenalin, a nadále pacienta monitorovat. Při ústupu symptomů lze zvážit pokračování infuze čtvrtinovou až poloviční rychlostí. Celkově se však jedná o léčbu dobře tolerovanou, se závažnými nežádoucími účinky se setkáváme vzácně [10,11]. Problémem zejména u dětských pacientů s MPS může být vzhledem ke zbytnělému podkoží zajištění žilního vstupu. Ve většině případů proto indikujeme zavedení venózního podkožního portu.
nežádoucích, zejména alergických reakcí (tab. 2) podává během hospitalizace. Infekt doprovázený horečkou nebo afekce, která může imitovat nežádoucí účinek, např. nově vzniklá vyrážka, je kontraindikací podání přípravku. Na možnost nežádoucích účinků je však nutno být připraven po celou dobu podávání léčby. V takovém případě je nezbytné zastavit aplikaci, zhodnotit závažnost reakce, podat infuzi krystaloidů, dle tíže reakce podat antihistaminika, antipyretika, ev. kortikoidy a adrenalin, a nadále pacienta monitorovat. Při ústupu symptomů lze zvážit pokračování infuze čtvrtinovou až poloviční rychlostí. Celkově se však jedná o léčbu dobře tolerovanou, se závažnými nežádoucími účinky se setkáváme vzácně [10,11]. Problémem zejména u dětských pacientů s MPS může být vzhledem ke zbytnělému podkoží zajištění žilního vstupu. Ve většině případů proto indikujeme zavedení venózního podkožního portu.
Účinek a limity ERT
V roce 2004 byla publikována první mezinárodní multicentrická studie zkoumající účinek ERT ve skupině 45 pacientů s MPS I léčených rekombinantně připravenou laronidázou po dobu 26 týdnů. V terapeutické skupině bylo oproti placebu pozorováno zvýšení forsírované vitální kapacity (forced vital capacity, FVC) o 5,6 % a zlepšení výsledku šestiminutového testu chůzí (6MWT) o 38,1 metru. Došlo k výraznému zmenšení hepatomegalie a ke sníženému vylučování glykosaminoglykanů v moči, u více postižených pacientů pak ke zmírnění spánkové apnoe/hypopnoe a ke zlepšení pohyblivosti v ramenním kloubu [23]. Do první dvojitě zaslepené studie s ERT u nemocných trpících MPS II bylo zařazeno 96 pacientů ve věku 5–31 let, kterým byla podávána idursulfáza jedenkrát týdně, ob týden nebo užívali placebo. V první skupině došlo po 53 týdnech podávání idursulfázy jednou týdně ke zvýšení výkonnosti vyjádřené pomocí 6MWT (průměr 37 metrů, p = 0,013) a ke zvýšení celkové FVC (průměr 160 ml, p = 0,001) ve srovnání s placebem [24]. Jednoznačný účinek byl prokázán též u nemocných s MPS VI – po pětiletém sledování došlo u 56 léčených pacientů k výraznému snížení vylučování glykosaminoglykanů (71–79 %), ke zlepšení v 12minutovém testu chůzí (průměr 183 metrů) [25]. Účinek léčby u MPS typu II byl jasně doložen při sledování 788 pacientů v mezinárodním registru (Hunter Outcome Score, HOS). U léčených ve srovnání s neléčenými pacienty byla prokázána delší doba přežití [26]. Účinek léčby není jednoznačný u MPS IV A, kde byly pozorované změny minimální [27]. Důvodem je zřejmě omezené působení ERT na rozvinuté kostní změny, které v klinickém obraze MPS IV A dominují (viz dále). Možno shrnout, že enzymová substituční terapie u pacientů s MPS zpomaluje progresi onemocnění, zlepšuje funkci postižených orgánů, mírní celou řadu příznaků a zlepšuje kvalitu života pacientů (tab. 1) [24,28]. Ovlivněny jsou zejména parenchymatózní a dobře vaskularizované, pro enzym přístupné orgány, tedy játra a slezina. Stabilizují se plicní funkce, zpomalena je progrese srdečního postižení. Kombinovaným účinkem pak dochází ke zvýšení svalové síly a výdrže monitorované šestiminutovým testem chůzí.
Přes nepochybný přínos ERT u pacientů s MPS je potřeba zmínit i její zásadní omezení. Rekombinantní enzym jako velká molekula nepřestupuje přes hematoencefalickou bariéru. Intravenózně podávaná ERT tak nemá vliv na postižení CNS [29]. V současné době jsou realizovány klinické studie s intratekální aplikací enzymu u pacientů s mukopolysacharidózou typu I, II a III A (www.clinicaltrials.gov) [30]. Paralelně s nimi probíhají studie zaměřené na využití endogenních receptorových systémů hematoencefalické bariéry mozku (blood brain barrier, BBB), které by umožnily transport terapeuticky účinného enzymu za využití dvou hlavních transportních systémů BBB – inzulinového a LRP‑1 (lipoprotein receptor‑related protein) [31].
Enzymová substituční terapie je rovněž špatně účinná v oblasti postižení srdečních chlopní, chrupavky a skeletu. Příčinou je nejspíše nedostatečná vaskularizace a časně vyjádřené změny postižených tkání s řadou výše zmíněných sekundárních patofyziologických mechanismů komplexně alterujících metabolismus buňky. Již přítomné postižení chlopní i kosterního aparátu (dysostosis multiplex) je pravděpodobně ireverzibilní a účinek ERT je nanejvýš stabilizující [32,33]. Zkouší se zvyšování biologické dostupnosti léčiva zvýšením podávané dávky či použitím modifikovaných molekul přípravku s vyšším biologickým poločasem eliminace. Další testovanou alternativou je využití chimérických enzymových molekul, které pak vstupují do buňky i cestou jiných než manózových či manóza‑6‑fosfátových receptorů, případně absorpční endocytózou. Vyvíjeny jsou nové aplikační cesty (např. intraartikulární aplikace) a biochemicky upravené přípravky s delším biologickým poločasem eliminace. [34].
Vzhledem k tomu, že endogenně produkovaný defektní protein může mít změněnou strukturu nebo vůbec není organismem vyráběn, může exogenně podávaná molekula proteinu vyvolávat v rámci ERT imunitní reakci s tvorbou protilátek, které mohou podávaný enzym inaktivovat. Míra této reakce je závislá mimo jiné na antigenicitě daného proteinu, na vlastnostech imunitního systému jedince či na genotypu jedince pro dané onemocnění. Na rozdíl od některých dalších lyzosomálních onemocnění (např. Pompeho nemoci) však nebývá klinický dopad přítomných protilátek u MPS klinicky významný [35].
Problematické je ukončování nebo přerušení ERT spojené s výrazným klinickým i laboratorním zhoršením. Objevují se závažné pneumonie, často s respiračním selháním, dochází k regresu vývoje, k rozvoji hydrocefalu, ke zhoršení očního a sluchového kontaktu, je přítomna svalová slabost, problémy s polykáním, retence hlenu v dýchacích cestách, otoky, zhoršení fyzické výdrže, změny na kůži či hepatomegalie [36,37]. Příčiny náhlého zhoršení zdravotního stavu pacientů po ukončení ERT nejsou doposud spolehlivě objasněny. Může se jednat o utlumení reziduální enzymové aktivity podávaným rekombinantním enzymem či o alterace procesu zpětné inhibice syntézy střádané makromolekuly v důsledku jejího hromadění, který se vyskytuje při přirozeném průběhu onemocnění.
Zásadní pro účinek léčby je časnost diagnostiky a zahájení léčby ideálně v presymptomatickém stadiu onemocnění. V takovém případě lze mnoha patologickým orgánovým projevům dokonce zcela předejít. Ilustrují to studie na sourozeneckých párech, kdy byl mladší sourozenec diagnostikován a léčen v presymptomatickém stadiu na základě diagnózy stanovené u staršího sourozence [38,39]. Lze tak usuzovat, že u symptomatických pacientů s MPS enzymová substituční terapie klinické projevy stabilizuje nebo zpomaluje jejich progresi, v presymptomatickém stadiu jim často dokáže zcela zabránit.
Časná iniciace ERT však vyžaduje určit diagnózu před samotnou manifestací onemocnění, což lze ve větším měřítku splnit jedině novorozeneckým screeningem. Kromě etických, technických a finančních problémů, které by takový screening přinášel, je největším problémem nízká genotypově‑fenotypová korelace. Řada mutací může mít nízkou penetranci, někteří pacienti mohou být postiženi atenuovanou formou onemocnění projevující se minimálními příznaky až v dospělosti. Je proto snaha hledat biomarkery ke stanovení přirozeného průběhu a prognózy onemocnění [40].
Rozhodnutí začít podávat ERT musí být vždy pečlivě zváženo zejména u pacientů se závažným fenotypem onemocnění. Enzymová substituční terapie představuje nezanedbatelnou zátěž pro pacienty, ale i pro celý zdravotnický systém. Jedná se o mimořádně finančně náročnou léčbu, která podle přípravku a tělesné hmotnosti pacienta vychází na jeden až dvanáct milionů korun ročně. O zahájení léčby proto rozhoduje lékařské konzilium na základě přesných indikačních kritérií. Účinek léčby je jednou ročně zhodnocen, posuzuje se přínos jejího pokračování.
Symptomatická léčba u pacientů s MPS
Významnou roli v péči o pacienty s MPS má symptomatická léčba. Fyzioterapie zahrnuje dechovou rehabilitaci, kterou se snažíme předcházet snížené pohyblivosti deformovaného hrudníku a udržet dostatečnou dechovou rezervu. Při pohybové rehabilitaci je nutné se zaměřit na postižení a stabilitu páteře (kyfóza, skolióza, hyperlordóza). Aktivním i pasivním cvičením je nutno předcházet vzniku kontraktur velkých i malých kloubů a později tyto deformity léčit. K udržení jemné motoriky je potřeba myslet i na prstová cvičení. Chirurgické zákroky si nejčastěji vyžádají operace tříselných a pupečních kýl. Pro opakované záněty středouší nebo kvůli syndromu spánkové obstrukční apnoe bývá indikována adenotomie, tonsilektomie, případně zavedení gromet. Z ortopedických výkonů to jsou korekční operace a implantace endoprotéz. Ve spolupráci s neurochirurgem je někdy nutno řešit útlak krční míchy, hydrocefalus, často je indikována operace syndromu karpálního tunelu. Je nutno zmínit významné anesteziologické riziko se ztíženou intubací (makroglossie, krátký krk, zúžení trachey apod.). Vzhledem k atlantoaxiální instabilitě je přísně kontraindikován záklon hlavy, doporučuje se zvážit endoskopicky asistovanou intubaci nosem. Zcela zásadní je psychoterapeutická péče. Péče o pacienty s MPS je velice náročná a vyžaduje maximální sociální a psychoterapeutickou podporu rodiny. Tam, kde je to možné, je vhodné snažit se pacienty integrovat mezi zdravé děti s individuálním pedagogickým přístupem.
Zatím jedinou a nejúčinnější prevencí dalšího výskytu MPS v postižených rodinách je prenatální diagnostika na základě stanovení aktivity enzymu v nativních a kultivovaných choriových klcích nebo v kultivovaných amniocytech a ověření přítomnosti patogenní mutace. Další možností je preimplantační genetická diagnostika. Podmínkou pro provedení prenatální diagnostiky je však včasné rozpoznání klinických příznaků a potvrzení diagnózy na úrovni enzymu a DNA.
Závěr
Enzymová substituční terapie zásadně změnila přirozený průběh onemocnění pacientů s mukopolysacharidózou. Zlepšuje kvalitu jejich života a prognózu jejich onemocnění. Pro maximální účinek enzymové substituční terapie je důležitá časná diagnostika a časné zahájení léčby. Jedná se o léčbu náročnou, u které je vždy nezbytné zohledňovat její omezení a rizika.
Práce byla podpořena projektem PRVOUK‑P24/LF1/3 a RVO‑VFN 64165/2012.
Seznam použité literatury
- [1] te Vruchte D, Wallom KL, Platt FM. Measuring relative lysosomal volume for monitoring lysosomal storage diseases. Methods Cell Biol 2015; 126: 331–347.
- [2] Neufeld EF, Muenzer J. The mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic & Molecular Bases of Inherited Disease, 8th edition vol. 3. McGraw Hill, New York, NY, USA. 2001.
- [3] Ballabio A, Giesermann V. Lysosomal disorders: From storage to cellular damage. Biochim Biophys Acta 2009; 1793: 684–696.
- [4] Poupětová H, Ledvinová J, Berná L, et al. The birth prevalence of lysosomal storage disorders in the Czech Republic: comparison with data in different populations. J Inherit Metab Dis 2010; 33: 387–396.
- [5] Muenzer J. The mucopolysaccharidoses: a heterogeneous group of disorders with variable pediatric presentations. J Pediatr 2004; 144: S27–34.
- [6] Ješina P, Magner M, Poupětová H, et al. Mukopolysacharidóza I – klinické projevy u 24 dětí z České republiky a Slovenska. Čes slov Pediat 2011; 66: 6–11.
- [7] Magner M, Hrubá E, Poupětová H, et al. Klinická manifestace Hunterovy nemoci u 22 českých pacientů. Čes slov Pediat 2014; 69 (S1): 56.
- [8] Burton BK, Giugliani R. Diagnosing Hunter syndrome in pediatric practice: practical considerations and common pitfalls. Eur J Pediatr 2012; 171: 631–639.
- [9] Solanki GA, Martin KW, Theroux MC, et al. Spinal involvement in mucopolysaccharidosis IVA (Morquio Brailsford or Morquio A syndrome): presentation, diagnosis and management. J Inherit Metab Dis 2013; 36: 339–355.
- [10] Kulhánek J, Albrecht J, Honzík T, Magner M. Psychiatrická manifestace dědičných metabolických poruch. Čes Slov Psychiat 2015; 111: 295–305.
- [11] Malinová V, Honzík T. Lysosomální onemocnění – současné možnosti diagnostiky a terapie. Pediatr praxi 2013; 14: 99–103.
- [12] Hers HG. Alpha glucosidase deficiency in generalized glycogenstorage disease (Pompe‘s disease). Biochem J 1963; 86: 11–16.
- [13] de Duve C. From cytases to lysosomes. Fed Proc 1964; 23: 1045–1049.
- [14] Fratantoni JC, Hall CW, Neufeld EF. Hurler and Hunter syndromes: mutual correction of the defect in cultured fibroblasts. Science 1968; 162: 570–572.
- [15] Coutinho MF, Matos L, Alves S. From bedside to cell biology: a century of history on lysosomal dysfunction. Gene 2015; 555: 50–58.
- [16] Ghosh P, Dahms NM, Korngeld S. Mannose 6 phosphate receptors: new twist in the tale. Nat Rev Mol Cell Biol 2003; 4: 202–212.
- [17] Barton NW, Brady RO, Dambrosia JM, et al. Replacement therapy for inherited enzyme deficiency – macrophage targeted glucocerebrosidase for Gaucher’s disease. N Engl J Med 1991; 324: 1464–1470.
- [18] Grabowski GA, Barton NW, Pastores G, et al. Enzyme therapy in type 1 Gaucher disease: comparative efficacy of mannose terminated glucocerebrosidase from natural and recombinant sources. Ann Intern Med 1995; 122: 33–39.
- [19] Ioannou YA, Bishop DF, Desnick RJ. Overexpression of human α galactosidase. A results in its intracellular aggregation, crystallization in lysosomes, and selective secretion. J Cell Biol 1992; 119: 1137–1150.
- [20] Kakkis ED, Matynia A, Jonas AJ, et al. Overexpression of the human lysosomal enzyme α l iduronidase in Chinese hamster ovary cells. Protein Expr Purif 1994; 5: 225–232.
- [21] Van Hove JL, Yang HW, Wu JY, et al. High level production of recombinant human lysosomal acid α glucosidase in Chinese hamster ovary cells which targets to heart muscle and corrects glycogen accumulation in fibroblasts from patients with Pompe disease. Proc Natl Acad Sci USA 1996; 93: 65–70.
- [22] Kulhánek J, Malinová V, Honzík T, Magner M. Enzymová substituční terapie u lysosomálních onemocnění. Čes slov Pediat 2015; 70: 224–231.
- [23] Wraith JE, Clarke LA, Beck M, et al. Enzyme replacement therapy for mucopolysaccharidosis I: a randomized, double blinded, placebo controlled, multinational study of recombinant human alpha
- [24] Muenzer J, Wraith JE, Beck M, et al. A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II (Hunter syndrome). Genet Med 2006; 8: 465–473.
- [25] Harmatz P, Giugliani R, Schwartz IV, et al. Long term follow up of endurance and safety outcomes during enzyme replacement therapy for mucopolysaccharidosis VI: Final results of three clinical studies of recombinant human N acetylgalactosamine 4 sulfatase. Mol Genet Metab 2008; 94: 469–475.
- [26] Burton BK, Jego V, Jones SA. Survival in idursulfase treated and untreated patients with MPS II. 12th Annual WORLDSymposium™, 29 February–4 March 2016, San Diego, CA, USA.
- [27] Hendriksz CJ, Burton B, Fleming TR, et al. Efficacy and safety of enzyme replacement therapy with BMN 110 (elosulfase alfa) for Morquio A syndrome (mucopolysaccharidosis IVA): a phase 3 randomised placebo controlled study. J Inherit Metab Dis 2014; 37: 979–990.
- [28] Muenzer J, Beck M, Eng CM, et al. Long term, open labeled extension study of idursulfase in the treatment of Hunter syndrome. Genet Med 2011; 13: 95–101.
- [29] Baldo G, Giugliani R, Matte U. Lysosomal enzymes may cross the blood brain barrier by pinocytosis: implications for enzyme replacement therapy. Med Hypotheses 2014; 82: 478–480.
- [30] Muenzer J, Hendriksz CJ, Fan Z, et al. A phase I/II study of intrathecal idursulfase IT in children with severe mucopolysaccharidosis II. Genet Med 2016; 18: 73–81.
- [31] Boado RJ, Zhang Y, Zhang Y, et al. Genetic engineering of a lysosomal enzyme fusion protein for targeted delivery across the human blood brain barrier. Biotechnol Bioeng 2008; 99: 475–484.
- [32] Muenzer J. Early initiation of enzyme replacement therapy for mucopolysaccharidoses. Mol Genet Metab 2014; 111: 63–72.
- [33] Braunlin E, Rosenfeld H, Kampmann C, et al. Enzyme replacement therapy for mucopolysaccharidosis VI: long term cardiac effects of galsulfase (Naglazyme®) therapy. J Inherit Metab Dis 2013; 36: 385–394.
- [34] Ortolano S, Viéitez I, Navarro C, et al. Treatment of lysosomal storage diseases: recent patents and future strategies. Recent Pat Endocr Metab Immune Drug Discov 2014; 8: 9–25.
- [35] Barbier AJ, Bielefeld B, Whiteman DA, et al. The relationship between anti idursulfase antibody status and safety and efficacy outcomes in attenuated mucopolysaccharidosis II patients aged 5 years and older treated with intravenous idursulfase. Mol Genet Metab 2013; 110: 303–310.
- [36] Jurecka A, Malinova V, Tylki Szymańska A. Effect of rapid cessation of enzyme replacement therapy: a report of 5 more cases. Mol Genet Metab 2014; 111: 212–213.
- [37] Jurecka A, Żuberuber Z, Opoka Winiarska V, et al. Effect of rapid cessation of enzyme replacement therapy: a report of 5 cases and a review of the literature. Mol Genet Metab 2012; 107: 508–512.
- [38] Tajima G, Sakura N, Kosuga M, et al. Effects of idursulfase enzyme replacement therapy for Mucopolysaccharidosis type II when started in early infancy: comparison in two siblings. Mol Genet Metab 2013; 108: 172–177.
- [39] Tylki Szymańska A, Jurecka A, Zuber Z, et al. Enzyme replacement therapy for mucopolysaccharidosis II from 3 months of age: a 3 year follow up. Acta Paediatr 2012; 101: e42–47.
- [40] Magner M, Kulhanek J, Ujcikova H, et al. Disorder in the house: actin level decrease in leukocytes of patients with Hunter syndrome. J Inherit Metab Dis 2015; Suppl 1 (38): S244.