Gaucherova nemoc a lysosomální onemocnění – současné možnosti diagnostiky a léčby
Souhrn:
Lysosomální střádavá onemocnění jsou vzácné, dědičně podmíněné nemoci způsobené nedostatečnou aktivitou některého z lysosomálních enzymů, transportních proteinů či kofaktorů. První příznaky se mohou objevit časně po narození i v pozdní dospělosti, časné formy mívají těžký průběh s rychlou progresí a s infaustní prognózou. Onemocnění je multisystémové s trvalou progresí obtíží a s postižením různých orgánů či tkání. Nejčastěji jde o metabolicky aktivní orgány či tkáně – centrální nervový systém, játra, myokard, svalstvo. Diagnózu definitivně potvrdíme průkazem nízké aktivity daného enzymu a/nebo molekulárně genetickým vyšetřením. Některé ze střádavých nemocí můžeme účinně léčit podáním rekombinantních enzymů formou pravidelných nitrožilních infuzí či omezením množství střádaného substrátu. U malého počtu pacientů s lysosomálním střádavým onemocněním je úspěšná transplantace kostní dřeně. Vzhledem k multisystémovému postižení je nutná mezioborová spolupráce včetně zajištění genetického poradenství a prenatální diagnostiky v rodinách pacientů. V současné době je diagnostikováno zhruba 70 různých typů lysosomálních střádavých onemocnění, z nichž k nejdéle známým patří i Gaucherova nemoc. Při Gaucherově nemoci dochází k lysosomálnímu střádání glukocerebrosidu v buňkách makrofágového původu. Střádání je nejvíce vyjádřeno v retikuloendotelu sleziny, kostní dřeně a v parenchymu jater. Dostupnou specifickou léčbu představuje podávání rekombinantních enzymů se srovnatelným a velice dobrým účinkem.
Key words: lysosomal disorders – Gaucher disease – enzyme replacement therapy – substrate reduction therapy.
Summary:
Lysosomal storage disorders are rare, hereditary illnesses caused by defective activity of some of the lysosomal enzymes, transport proteins or cofactors. The first signs can appear both soon after birth and in late adulthood, early onset forms being often severe, with rapid progression and hopeless prognosis. The disease is a multisystem one and is characterized by permanent progression of symptoms and by affection of many organs and tissues. Most often, metabolically active organs and tissues are affected, such as the central nervous system, liver, myocardium, and muscles. The diagnosis can be confirmed by demonstrating low activity of a given enzyme and/or by molecular genetic evaluation. Some of the storage diseases can be treated effectively with recombinant enzymes applied intravenously on a regular basis or by reducing the amount of stored substrate. A small number of patients with lysosomal storage disorders can profit from bone marrow transplants. Given the multisystem nature of the disease, interdisciplinary approach is warranted, including genetic counseling and prenatal evaluation in the patients’ families. Currently, about 70 types of lysosomal storage disorders are being diagnosed, Gaucher disease being one of those known for the longest time period. Gaucher disease is characterized by lysosomal storage of glucocerebroside in the cells of macrophageal origin. Storage is pronounced the most in the reticuloendothelial system of the spleen and bone marrow and in the liver parenchyma. Specific therapy is available, consisting of the application of recombinant enzymes with comparable and very favorable effect.
Úvod
Lysosomální onemocnění patří mezi dědičné metabolické poruchy. Na klinické, enzymatické a molekulární úrovni bylo popsáno asi 70 různých lysosomálních poruch. Dědičnost je většinou autosomálně recesivní, výjimkou je Fabryho nemoc a mukopolysacharidóza II s typem dědičnosti vázaným na chromosom X. Jedná se o vzácná onemocnění vyskytující se panetnicky, jejich sumární ![Graf 1 Podíl zastoupení jednotlivých typů lysosomálních střádavých onemocnění v České republice; podle [1] – Poupetova, et al., 2010. GP – glykoproteinóza; PN – Pompeho nemoc; ML – mukolipidóza; MPS – mukopolysacharidóza; NCL – neuronální ceroidní lipofuscinóza](https://www.remedia.cz/photo-a-30700---.jpg) výskyt v naší populaci je odhadován poměrem 1 : 8 200 živě narozených dětí [1,2].
výskyt v naší populaci je odhadován poměrem 1 : 8 200 živě narozených dětí [1,2].
Podíl zastoupení jednotlivých typů a diagnóz lysosomálních střádavých onemocnění v České republice uvádí graf 1, 2 [1].
Gaucherova nemoc
Gaucherova nemoc (Gaucher disease, GD) je dědičná porucha metabolismu s autosomálně recesivním typem dědičnosti, při které dochází k lysosomálnímu střádání glukocerebrosidu (glukosylceramidu) v buňkách makrofágového původu. Onemocnění je způsobeno nedostatečnou aktivitou β‑glukocerebrosidázy (β‑glukosidázy), lysosomálního enzymu, který štěpí glukocerebrosid na glukózu a ceramid. Podle klinického průběhu onemocnění se Gaucherova nemoc dělí na tři typy: non‑neuronopatický typ I, akutní neuronopatický typ II a subakutní neuronopatický typ III.![Graf 2 Podíl zastoupení jednotlivých diagnóz lysosomálních střádavých onemocnění v České republice; podle [1] – Poupetova, et al., 2010. CESD – cholesterol ester storage disease, nemoc ze střádání cholesterolu; GM1, GM2 – gangliosidózy; MLD – metachromatická leukodystrofie; NP A, B, C – Niemannova–Pickova nemoc typu A, B, C](https://www.remedia.cz/photo-a-30701---.jpg)
K typickým projevům onemocnění patří hepatosplenomegalie, bolesti kostí, zvýšená krvácivost, u dětí menší vzrůst a neprospívání, u II. a III. typu i postižení centrálního nervového systému (CNS). Při laboratorním vyšetření nacházíme snížený počet trombocytů, nižší koncentraci hemoglobinu, častá je hypergamaglobulinemie. Diagnózu potvrdí nízká aktivita β‑glukocerebrosidázy a nález specifické mutace v genu GBA. Metodu první léčebné volby představuje enzymatická substituční terapie. Vzhledem k postižení více systémů je nutná spolupráce se specialisty řady dalších oborů (kardiologie, neurologie, oční lékařství, otorinolaryngologie, ortopedie, revmatologie, rehabilitace), symptomatická terapie a komplexní péče včetně genetického poradenství a prenatální diagnostiky.
Epidemiologie
Gaucherova nemoc se vyskytuje panetnicky s prevalencí udávanou v rozpětí 1 : 57 000 až 1 : 100 000 živě narozených dětí. V České republice byla prevalence pro Gaucherovu nemoc všech typů stanovena poměrem 1 : 88 000, pro heterozygoty poměrem 1 : 149 [1]. Vyšší výskyt byl popsán v uzavřených etnických populacích – v židovské populaci Ashkenazi byla zjištěna prevalence 1 : 1 000 s frekvencí heterozygotů 1 : 14.
Ve více než 90 % případů se vyskytuje Gaucherova nemoc I. typu. Pouze 5–10 % z celkového počtu pacientů trpí variantou s postižením nervového systému – tedy II. nebo III. typem. Vyšší výskyt III. typu byl dokumentován v populaci severního Švédska (tzv. Norrbottnian typ GD III) [3,4], v Polsku a v části arabské populace (Jenin‑Arab).
Historie
Onemocnění poprvé popsal lékař Ernest Phillipe Gaucher roku 1882 a charakterizoval je jako významnou splenomegalii nenádorového charakteru. V letech 1901–1912 bylo vypozorováno, že toto onemocnění postihuje více systémů současně a má familiární výskyt. V roce 1920 bylo poprvé identifikováno prudce probíhající neurologické postižení u infantilní formy, které bylo označeno jako Gaucherova nemoc typu II. V roce 1934 byla identifikována látka, která je střádána, a byla označena jako glukocerebrosid. Typ dědičnosti byl popsán v roce 1950 (autosomálně recesivní dědičnost – Herndon, Bender). V roce 1958 si Fried všiml mnohonásobně vyšší incidence tohoto onemocnění v židovské populaci Ashkenazi a o rok později popsal Hillborg smíšený typ III se závažným viscerálním a pomalu progredujícím postižením CNS. V roce 1965 byl identifikován příslušný enzym – β‑glukocerebrosidáza (Brady), od roku 1972 byla možná prenatální diagnostika. V roce 1985 byla klonována celodélková cDNA pro glukocerebrosidázu a byla popsána genetická heterogenita onemocnění. První klinický pokus o enzymatickou substituční terapii aglucerázou proběhl úspěšně v roce 1991 ve Spojených státech amerických (Barton, Brady), v České republice pak v roce 1995 (Zeman). Léčba byla zpočátku prováděna enzymem extrahovaným z placent, později byla nahrazena podáním rekombinantního enzymu imiglucerázy.
Etiologie a patogeneze
Gaucherova nemoc je jednou z nejčastějších poruch katabolismu glykosfingolipidů. Nedostatečná tvorba či tvorba vadného lysosomálního enzymu β‑glukocerebrosidázy způsobuje snížené štěpení glukocerebrosidu, metabolického meziproduktu degradace glykosfingolipidů, který je za normálních okolností štěpen na glukózu a ceramid. ![Obr. 1 Schéma metabolismu glukocerebrosidu; podle [3] – Conradi, et al., 1984.](https://www.remedia.cz/photo-a-30702---.jpg) Glukocerebrosid je podstatnou součástí buněčných membrán. Po zániku buněk jsou jednotlivé součásti buněk štěpeny a reutilizovány. Největším zdrojem glukocerebrosidu jsou červené a bílé krvinky, jejichž životní cyklus trvá 20–30 dnů. Při nedostatečné aktivitě β‑glukocerebrosidázy dochází k hromadění velkého množství glukocerebrosidu nad enzymatickým blokem (obr. 1). Nedegradovaný substrát je vychytáván buňkami monocyto‑makrofágového systému a hromadí se v lysosomech různých tkání či orgánů. Většinou se jedná o metabolicky aktivní orgány, z nichž nejdříve bývá postižena kostní dřeň, poté retikuloendotel sleziny, Kuppferovy buňky v játrech, osteoklasty. Postižen může být kterýkoli orgán či tkáň včetně CNS, plic, oka (sklivec) [5].
Glukocerebrosid je podstatnou součástí buněčných membrán. Po zániku buněk jsou jednotlivé součásti buněk štěpeny a reutilizovány. Největším zdrojem glukocerebrosidu jsou červené a bílé krvinky, jejichž životní cyklus trvá 20–30 dnů. Při nedostatečné aktivitě β‑glukocerebrosidázy dochází k hromadění velkého množství glukocerebrosidu nad enzymatickým blokem (obr. 1). Nedegradovaný substrát je vychytáván buňkami monocyto‑makrofágového systému a hromadí se v lysosomech různých tkání či orgánů. Většinou se jedná o metabolicky aktivní orgány, z nichž nejdříve bývá postižena kostní dřeň, poté retikuloendotel sleziny, Kuppferovy buňky v játrech, osteoklasty. Postižen může být kterýkoli orgán či tkáň včetně CNS, plic, oka (sklivec) [5].
Onemocnění je autosomálně recesivně dědičné, gen pro β‑glukocerebrosidázu je situován na 1. chromosomu (GBA, 1q21) a bylo v něm popsáno více než 400 mutací (databáze HGMD, The Human Gene Mutation Database). V České republice patří k nejčastějším mutace p.N370S, která je v homozygotním stavu spojována spíše s lehčím průběhem onemocnění, a mutace p.L444P, která je naopak typická pro těžší průběh s postižením CNS [3,4,6]. Korelace fenotypu s genotypem či se zbytkovou aktivitou enzymu však není absolutní.
Původní mechanistická teorie předpokládala alteraci funkce postižené buňky v důsledku nahromadění glukocerebrosidu v lysosomu. Pozdější poznatky prokázaly, že jde o komplexní proces s narušením buněčné homeostázy vedoucí až k plánované buněčné smrti – apoptóze.
Aktivované makrofágy vylučují zvýšené množství cytokinů jako interleukinů IL‑6, IL‑10, IL‑1β, dále tumor nekrotizující faktor alfa (TNFα) a růstový faktor pro makrofágy (macrophage colony stimulating factor, M‑CSF) [7,8], které se uplatňují při rozvoji zánětu; IL‑6 stimuluje kostní resorpci a tlumí novotvorbu kosti inhibicí sekreční aktivity osteoblastů (mineralizace). Zvýšená aktivita IL‑6 může být jednou z příčin často pozorované osteopenie či osteoporózy u pacientů s Gaucherovou nemocí v mladém věku. Současně dochází ke zvýšené expresi povrchových receptorů na buňkách imunitního systému – makrofágů, buněk T, B a NK (natural killers, přirození zabíječi). Aktivace makrofágů vede ke změně buněčné biologie včetně abnormálních intercelulárních či intracelulárních signálů. Chronická stimulace imunitního systému může mít za následek klonální expanzi buněk B s častým rozvojem polyklonální či monoklonální gamapatie, MGUS (monoclonal gammopathy of undetermined significance, monoklonální gamapatie nejasného významu) a myelomu [9].
Patofyziologie postižení CNS u neuronopatických forem Gaucherovy nemoci není dosud zcela objasněna. Zvažuje se možnost poškození neuronů glukocerebrosidem, který cirkuluje v krevním oběhu a je následně transportován a hromaděn v mozku. Jiná z teorií předpokládá, že k poškození CNS dochází působením jiného glykosfingolipidu – glukosylsfingosinu (deacetylovaného analoga glukocerebrosidu), k jehož degradaci je nutný stejný enzym – β‑glukocerebrosidáza. Glukosylsfingosin je zastoupen v minimálním množství, tvoří přibližně 1 % nastřádaného glukocerebrosidu, je však značně neurotoxický [6].
Klinický obraz
Podle orgánového postižení a věku pacienta v době rozvoje prvních příznaků onemocnění rozdělujeme schematicky Gaucherovu nemoc do tří základních typů (obr. 2). Spektrum klinických projevů je však heterogenní – 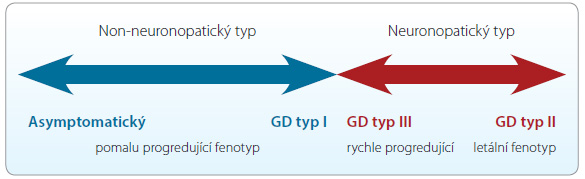 od asymptomatických forem přes pomalu progredující až po rychle progredující formy s fatálním průběhem v raném věku [10].
od asymptomatických forem přes pomalu progredující až po rychle progredující formy s fatálním průběhem v raném věku [10].
Typ I – non‑neuronopatický, adultní, viscerální
Typ I se vyskytuje nejčastěji, první příznaky se objevují většinou v pozdním dětství a kdykoli později v dospělosti. Pacienti si stěžují na únavu, na snadnou tvorbu modřin, na bolesti kostí či kloubů, na bolesti břicha. Příznaky krvácivé diatézy mohou být i ve formě protrahovaného krvácení po menších chirurgických výkonech, opakované epistaxe, u dívek může být silnější menstruace. Zvýšená krvácivost je způsobena útlakem červené kostní dřeně střádaným glukocerebrosidem s následným poklesem krvetvorby. První bývají postiženy trombocyty, poté erytrocyty a leukocyty.
Kostní krize
Nárůst střádání působí remodelaci kosti a útlak cévního zásobení [11]. Důsledkem jsou často intenzivní bolesti kostí, které mohou vyústit až v tzv. kostní krizi. Pacient trpí krutými bolestmi kostí, po několik dnů nebývá schopen a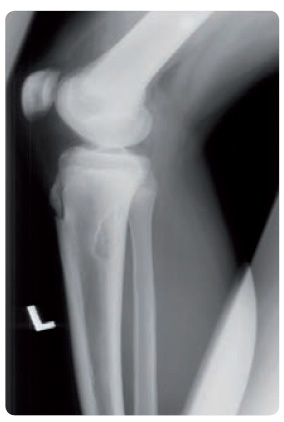 ktivního pohybu. Tento stav je často doprovázen celkovými příznaky včetně teploty, leukocytózy a vyšší zánětlivé aktivity. Kostní krize může být chybně diagnostikována jako akutní osteomyelitida, hemokultura je však opakovaně negativní. Postižení skeletu se manifestuje patologickými frakturami dlouhých kostí, žeber či obratlů v důsledku ztenčení kortikální kosti a osteoporózy. Časté bývají ohraničené lytické léze (obr. 3) či kostní infarkty. Postižení kloubů působí předčasné degenerativní změny až aseptickou nekrózu s následným rozpadem hlavice humeru či femuru. Časté jsou infekční komplikace – septické artritidy, osteomyelitidy.
ktivního pohybu. Tento stav je často doprovázen celkovými příznaky včetně teploty, leukocytózy a vyšší zánětlivé aktivity. Kostní krize může být chybně diagnostikována jako akutní osteomyelitida, hemokultura je však opakovaně negativní. Postižení skeletu se manifestuje patologickými frakturami dlouhých kostí, žeber či obratlů v důsledku ztenčení kortikální kosti a osteoporózy. Časté bývají ohraničené lytické léze (obr. 3) či kostní infarkty. Postižení kloubů působí předčasné degenerativní změny až aseptickou nekrózu s následným rozpadem hlavice humeru či femuru. Časté jsou infekční komplikace – septické artritidy, osteomyelitidy.
Hepatosplenomegalie
Střádání ve slezině působí několikanásobné zvětšení jejího objemu s útlakem nitrobřišních orgánů (obr. 4, 5). V parenchymu sleziny postupně dochází k nodulární přestavbě v důsledku ložiskové akumulace Gaucherových buněk či po proběhlém infarktu sleziny. Jeho projevem bývá náhlá krutá bolest břicha, může však proběhnout i asymptomaticky. Důsledkem střádání v játrech je většinou spíše zhoršení průtoku krve játry s postupným rozvojem portální hypertenze a vznikem portokaválních zkratů. Střádání je vyjádřeno v Kuppferových buňkách, hepatocyty bývají ušetřeny, jaterní funkce je bez větší alterace. V dětském věku je typickým příznakem hmotnostní neprospívání, nižší vzrůst a opožděný nástup puberty.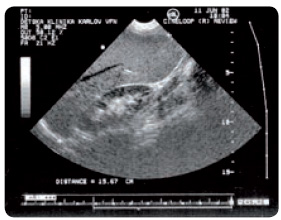
Komplikace
Rizikovým obdobím u žen je gravidita – u neléčených pacientek je popsán vyšší počet spontánních abortů, více komplikací v důsledku krvácení či septických stavů během porodu, v šesti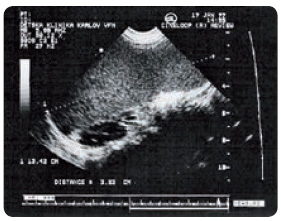 nedělí i v průběhu laktace [12].
nedělí i v průběhu laktace [12].
Adultní typ Gaucherovy nemoci byl dlouhodobě považován za selektivně viscerální postižení bez neurologických příznaků. Nevyskytuje se dysmorfie, mentální vývoj je bez patologie. V posledních letech byla věnována pozornost zvýšenému výskytu Parkinsonovy nemoci u pacientů s Gaucherovou nemocí I. typu. Prevalence Parkinsonovy nemoci v běžné populaci je 0,3 %, u pacientů ve věku nad 60 let dosahuje 1 %, prevalence v přítomnosti mutace spojené s výskytem Gaucherovy nemoci se zvyšuje na 9 %, riziko je větší i pro heterozygoty. První příznaky Parkinsonovy nemoci se u pacientů s Gaucherovou chorobou rozvíjejí v mladším věku, častý je familiární výskyt, farmakorezistence a rychlý rozvoj demence [13–16].
U pacientů s Gaucherovou nemocí je popisován zvýšený výskyt maligních onemocnění, zvláště mnohočetnéh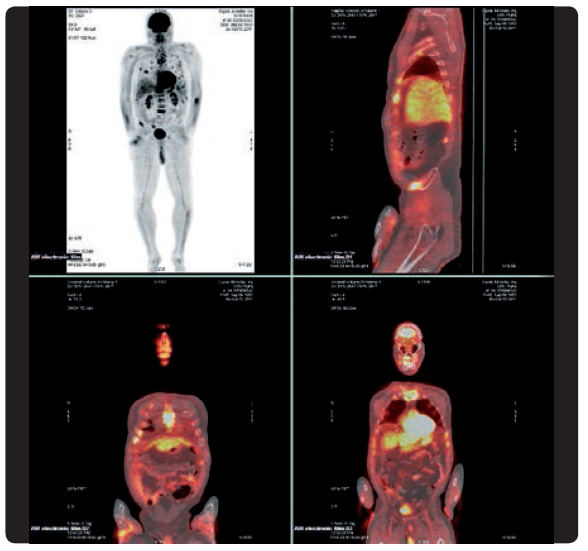 o myelomu [17], viz obr. 6. Vyšší je i riziko rozvoje dalších lymfoproliferativních onemocnění či solidních tumorů [18].
o myelomu [17], viz obr. 6. Vyšší je i riziko rozvoje dalších lymfoproliferativních onemocnění či solidních tumorů [18].
Další komplikací u pacientů s Gaucherovou nemocí může být rozvoj plicní hypertenze. Většinou postihuje pacienty po splenektomii s těžkým průběhem onemocnění, výskyt bývá familiární.
Typ II – akutní neuronopatický typ
Typ II je nejzávažnější formou Gaucherovy nemoci. Příznaky se objevují již prenatálně či perinatálně, nejpozději v prvních měsících života. V nejzávažnějším případě se narodí mrtvý plod s příznaky hydrops fetalis. K typickým příznakům patří masivní hepatosplenomegalie, kachexie, retroflexe krku, rychle progredující neurologické postižení ve smyslu bulbárního syndromu, zástavy vývoje, myoklonické epilepsie či atypických očních pohybů. Kožní postižení má většinou charakter generalizované kongenitální ichtyózy typu collodion baby. Až ve 35 % mohou být přítomny dysmorfické rysy. K úmrtí dochází většinou do věku dvou let [19].
Typ III – subakutní neuronopatický typ
Typ III je nejméně častý, většinou jde o kombinaci závažných viscerálních příznaků již v kojeneckém či batolecím věku (obr. 7) s pozdějším pomalu progredujícím neurologickým postižením. V předškolním či školním věku se mohou objevit atypické pohyby hlavy a očí, strabismus, nystagmus, sekundární epilepsie, myoklonické křeče, tremor, dystonie, zpomalení vývoje až pomalu progredující 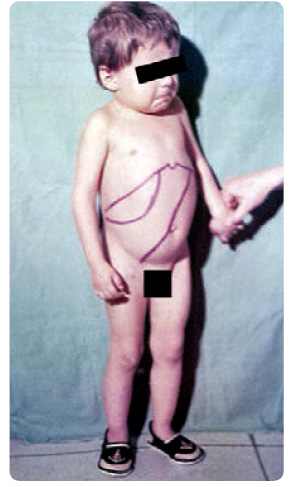 demence. U některých pacientů se postupně rozvíjí deformita hrudníku a páteře až gibus [3,4,6], u dalších je nejzávažnějším příznakem postižení myokardu, chlopní a aorty, často s masivními kalcifikacemi [20].
demence. U některých pacientů se postupně rozvíjí deformita hrudníku a páteře až gibus [3,4,6], u dalších je nejzávažnějším příznakem postižení myokardu, chlopní a aorty, často s masivními kalcifikacemi [20].
Diagnostika
Diagnózu může pomoci určit podrobná anamnéza, která potvrdí typické obtíže – únavu, zvýšenou krvácivost, bolesti kostí či kloubů. V klinickém nálezu se zaměříme na ověření hepatosplenomegalie (cave – slezina může zasahovat přes střední čáru či do malé pánve, dolní okraj sleziny nemusí být hmatný). V krevním obraze je typický pokles počtu trombocytů, nižší koncentrace hemoglobinu, případně leukopenie. V koagulačním vyšetření nacházíme často prodloužený aktivovaný parciální tromboplastinový čas (APTT), typické jsou vysoké hodnoty D‑dimerů. V biochemickém vyšetření může být patrná mírná elevace aktivity jaterních aminotransferáz, typický je nález vysokých koncentrací ferritinu či dyslipidemie. Dalším častým nálezem je hypergamaglobulinemie, odchylky v imunoelektroforéze mohou mít charakter monoklonální či polyklonální gamapatie [9].
Nespecificky podporuje diagnózu zjištění vysokých koncentrací biomarkerů. Jde o proteiny, jejichž aktivita se zvyšuje v případě aktivace monocyto‑makrofágového systému. Hodnoty biomarkerů korelují s aktivitou nemoci, jsou významnými pomocnými faktory při stanovení diagnózy střádavého onemocnění a pomáhají při kontrole účinnosti léčby. Pro diagnostiku Gaucherovy nemoci využíváme biomarkery chitotriosidázu, protein CCL18, tartarát rez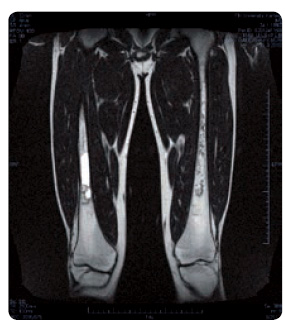 istentní kyselou fosfatázu a angiotensin konvertující enzym. Zvláště chitotriosidáza je citlivým markerem aktivity buněk makrofágového systému u Gaucherovy nemoci. Zároveň je nutno připomenout, že normální nebo nízká aktivita některého z biomarkerů nevylučuje diagnózu střádavého onemocnění (zhruba 5 % populace je deficitních pro chitotriosidázu). V našem souboru bylo v roce 2015 vyšetřeno 35 pacientů s Gaucherovou nemocí na přítomnost nejčastější mutace v genu CHIT1. Přímou sekvenací produktu polymerázové řetězové reakce zahrnujícího exon 10 byla prokázána přítomnost mutace c.1049_1072dup24 v heterozygotním stavu u 13 pacientů, tedy u 37 % pacientů ve vyšetřovaném souboru, u zbylých 21 vyšetřených pacientů nebyla přítomnost této mutace potvrzena.
istentní kyselou fosfatázu a angiotensin konvertující enzym. Zvláště chitotriosidáza je citlivým markerem aktivity buněk makrofágového systému u Gaucherovy nemoci. Zároveň je nutno připomenout, že normální nebo nízká aktivita některého z biomarkerů nevylučuje diagnózu střádavého onemocnění (zhruba 5 % populace je deficitních pro chitotriosidázu). V našem souboru bylo v roce 2015 vyšetřeno 35 pacientů s Gaucherovou nemocí na přítomnost nejčastější mutace v genu CHIT1. Přímou sekvenací produktu polymerázové řetězové reakce zahrnujícího exon 10 byla prokázána přítomnost mutace c.1049_1072dup24 v heterozygotním stavu u 13 pacientů, tedy u 37 % pacientů ve vyšetřovaném souboru, u zbylých 21 vyšetřených pacientů nebyla přítomnost této mutace potvrzena.
Velmi nápomocné jsou zobrazovací metody – ultrasonograficky lze prokázat zvětšení jater, sleziny, hyperechogenitu parenchymu obou orgánů a zaoblení dolního úhlu jater při střádání, ložiskové změny při nodulární přestavbě či při infarktech. Pro nepřímé známky portální hypertenze svědčí dilatace a obrácení toku krve ve vena liena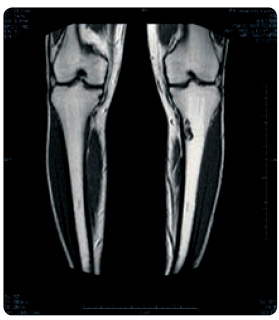 lis, v. portae a v. mesenterica sup. Na magnetické rezonanci (MR) lze zobrazit změnu signálu kostní dřeně v T1W v důsledku nahromadění glukocerebrosidu v dlouhých kostech, v tělech obratlů či v pánevních kostech (obr. 8, 9). Zralá kostní dřeň obsahuje za normálních okolností velké množství tukových látek a v T1W je patrný typický hyperintenzní signál. Nahromadění Gaucherových buněk v kostní dřeni způsobí redukci signálu odpovídající zhruba zobrazení svalové tkáně. Hodnocení nálezů MR u dětí je obtížnější, protože červená kostní dřeň s vysokou aktivitou hemopoezy obsahuje méně lipidů a její signál v T1W je nižší. Její náhrada zralou kostní dření s vyšším obsahem lipidů probíhá postupně v závislosti na věku. Někdy může být chybně vyhodnocen nález v oblasti fyziologicky přítomné červené kostní dřeně jako infiltrace Gaucherovými buňkami (obr. 10), naopak její náhrada tukovou kostní dření v rámci maturace může být falešně vyhodnocena jako pozitivní účinek léčby [21]. Vyšetření skeletu pomocí MR bývá prováděno opakovaně, proto mu dáváme přednost před jinými zobrazovacími metodami, jako je např. scintigrafie či výpočetní tomografie (computed tomography, CT), u který
lis, v. portae a v. mesenterica sup. Na magnetické rezonanci (MR) lze zobrazit změnu signálu kostní dřeně v T1W v důsledku nahromadění glukocerebrosidu v dlouhých kostech, v tělech obratlů či v pánevních kostech (obr. 8, 9). Zralá kostní dřeň obsahuje za normálních okolností velké množství tukových látek a v T1W je patrný typický hyperintenzní signál. Nahromadění Gaucherových buněk v kostní dřeni způsobí redukci signálu odpovídající zhruba zobrazení svalové tkáně. Hodnocení nálezů MR u dětí je obtížnější, protože červená kostní dřeň s vysokou aktivitou hemopoezy obsahuje méně lipidů a její signál v T1W je nižší. Její náhrada zralou kostní dření s vyšším obsahem lipidů probíhá postupně v závislosti na věku. Někdy může být chybně vyhodnocen nález v oblasti fyziologicky přítomné červené kostní dřeně jako infiltrace Gaucherovými buňkami (obr. 10), naopak její náhrada tukovou kostní dření v rámci maturace může být falešně vyhodnocena jako pozitivní účinek léčby [21]. Vyšetření skeletu pomocí MR bývá prováděno opakovaně, proto mu dáváme přednost před jinými zobrazovacími metodami, jako je např. scintigrafie či výpočetní tomografie (computed tomography, CT), u který![Obr. 10 Cytologický obraz lysosomálního střádání v makrofázích v nátěru kostní dřeně – Gaucherova buňka s charakteristickou cytoplazmou vzhledu „zmačkaného papíru“ s excentricky umístěným jádrem, PAS (Periodic Acid Schiff) barvení; podle [10] – Gaucher disease and Cerezyme® monograph, 2005.](https://www.remedia.cz/photo-a-30711---.jpg) ch je rizikem kumulativní radiační zátěž. Rentgenovým vyšetřením je možno hodnotit ztenčení kortikální kosti, přestavbu a remodelaci dlouhých kostí (obr. 3), případně dokumentovat patologické fraktury [5,22,23]. Nižší kostní denzita je častým nálezem, není však typická pouze pro diagnózu Gaucherovy nemoci. Definitivním potvrzením diagnózy Gaucherovy nemoci je průkaz nedostatečné aktivity β‑glukocerebrosidázy v izolovaných leukocytech periferní krve. Enzymologické vyšetření lze provádět i ve fibroblastech, v bioptických vzorcích tkání, v amniocytech či v buňkách choriových klků. Na molekulárně genetické úrovni pak diagnózu verifikujeme nálezem dvou mutantních alel genu GBA [24–26].
ch je rizikem kumulativní radiační zátěž. Rentgenovým vyšetřením je možno hodnotit ztenčení kortikální kosti, přestavbu a remodelaci dlouhých kostí (obr. 3), případně dokumentovat patologické fraktury [5,22,23]. Nižší kostní denzita je častým nálezem, není však typická pouze pro diagnózu Gaucherovy nemoci. Definitivním potvrzením diagnózy Gaucherovy nemoci je průkaz nedostatečné aktivity β‑glukocerebrosidázy v izolovaných leukocytech periferní krve. Enzymologické vyšetření lze provádět i ve fibroblastech, v bioptických vzorcích tkání, v amniocytech či v buňkách choriových klků. Na molekulárně genetické úrovni pak diagnózu verifikujeme nálezem dvou mutantních alel genu GBA [24–26].
Diferenciální diagnostika
Diferenciálně diagnosticky je nutno odlišit jiná onemocnění charakterizovaná podobnými příznaky. Jde převážně o lymfoproliferativní onemocnění (lymfom, leukemie, myelom), u kterých je vyjádřena splenomegalie a útlum kostní dřeně. Izolovaná splenomegalie může být způsobena chronickou infekcí jak bakteriální, tak virové (virus Epsteina–Barrové, cytomegalovirus) či protozoární etiologie (toxoplazmóza). Z hematologických příčin splenomegalie je možno zvažovat vrozenou či získanou hemolytickou anemii, případně extramedulární hematopoezu při talasemii, osteopetrózu či myelofibrózu. Slezina může být zvětšena v důsledku městnání krve při fibróze či cirhóze jater, při obstrukci portální či lienální žíly nebo při městnavém srdečním selhání. V diferenciálně diagnostické rozvaze je nutno uvést další střádavá onemocnění – lipidózy – Niemannovu–Pickovu nemoc, mukopolysacharidózy či infiltraci sleziny při histiocytóze. Splenomegalie se vyskytuje i při systémových onemocněních, jako je lupus erythematodes, sarkoidóza, revmatoidní artritida. Izolovaná trombocytopenie bývá nejčastěji autoimunitní etiologie. Protilátky proti trombocytům jsou však často pozitivní i u pacientů s Gaucherovou nemocí, přítomnost antitrombocytárních protilátek proto nevylučuje diagnózu Gaucherovy nemoci. Projevy krvácivé diatézy nacházíme u poruch hemokoagulace, hodnoty jednotlivých koagulačních faktorů jsou u Gaucherovy nemoci bez kritických odchylek. Kostní krizi lze snadno zaměnit za akutní osteomyelitidu, aseptická nekróza hlavice femuru pak může být v dětském věku mylně diagnostikována jako morbus Perthes. Bolesti kostí u dětí jsou často hodnoceny jako růstové bolesti. Hmotnostní neprospívání a menší vzrůst u dětí spolu s anemií jsou nejčastěji způsobeny malabsorpcí (celiakie). U dětí s Gaucherovou nemocí je neprospívání způsobeno útlakem gastrointestinálního traktu při organomegalii a předčasným pocitem plnosti. Jejich kalorický příjem je celkově nižší. Stejně tak opožděný nástup puberty není u pacientů s Gaucherovou nemocí způsoben odchylkami v endokrinním systému.
Léčba
Enzymatická substituční léčba
U nejčastějšího typu I (viscerálního) je zásadní včasné zahájení enzymatické substituční léčby (enzyme replacement therapy, ERT). V současné době je v ČR k dispozici imigluceráza (Cerezyme®) [2,5,10,27] a velagluceráza (Vpriv®) [28]. Enzymy jsou podávány ve dvouhodinových nitrožilních infuzích v intervalu dvou týdnů. Doporučená dávka je zhruba 30 j./kg/2 týdny, další úprava dávek je individuální dle tíže postižení, dle reakce na léčbu, dle klinického nálezu a laboratorních ukazatelů včetně biomarkerů. Vzhledem k tomu, že jde o aplikaci proteinového derivátu, je možno očekávat v rámci nežádoucích účinků alergickou reakci až anafylaktického typu. U našich pacientů s Gaucherovou nemocí I. a III. typu jsme závažnější komplikace v průběhu enzymatické léčby nezaznamenali. Do 6–12 měsíců po zahájení léčby je možno pozorovat ústup subjektivních obtíží, úpravu hematologických parametrů, do jednoho roku až dvou let zástavu progrese střádání a postižení skeletu. V případě II. a III. typu enzymatická substituční léčba zmírní viscerální příznaky, velká molekula enzymu však neproniká přes hematoencefalickou bariéru a neovlivní postižení CNS. Výhodou léčby je okamžitý účinek, minimální nežádoucí účinky, možnost podání u dětí, u gravidních pacientek i v průběhu laktace. Nevýhodou je nutnost intravenózní aplikace, nemožnost ovlivnit postižení CNS a vysoká cena.
Substrát redukující léčba
Další možností léčby je léčba založená na snížení syntézy substrátu (substrate reduction therapy, SRT). Ta vychází z principu deprivace substrátu, tudíž snížené množství glukocerebrosidu je možno zpracovat i při nízké aktivitě daného enzymu (β‑glukocerebrosidázy). Důsledkem je omezení střádání a navození rovnovážného stavu v buňce.
Jde o malé molekuly, které mají schopnost pronikat do CNS a teoreticky ovlivnit i neurologické postižení. V ČR je registrován pro léčbu pacientů s Gaucherovou nemocí iminocukr miglustat (Zavesca®) a generický přípravek Cromiva® [29]. Výhodou je možnost perorálního podání a nižší cena, nevýhodou pak nemožnost podat tento typ léčby dětem a pacientům ve fertilním věku i časté nežádoucí účinky – průjmy, bolesti břicha, tremor, dyskineze, neuropatie. V současnosti byla v klinických studiích ověřena účinnost další malé molekuly – eliglustatu (Cerdelga®) [30]. Princip působení eliglustatu spočívá ve specifické inhibici glukosylceramidsyntázy. V ČR je tento přípravek sice registrován, ale zatím není dostupný. Dle vyhodnocení klinických studií je účinek eliglustatu srovnatelný s enzymatickou substituční léčbou. Nutná je opatrnost u pacientů, kteří mají pomalý metabolismus zprostředkovaný CYP2D6 a eventuální postižení myokardu a kardiovaskulárního systému [31]. Dle charakteristiky genotypu CYP2D6 lze určit, zda je pacient rychlý, střední nebo pomalý metabolizátor. Podle nálezu je třeba upravit dávkování, vyvarovat se rizika současného podávání léčiv inhibujících CYP2D6 a CYP3A. Při vyšších koncentracích eliglustatu bylo popsáno prodloužení intervalu PR, QRS a/anebo QTc, které může vyústit v závažnou srdeční arytmii.
Chaperony
Léčba chaperony je dosud aplikována pouze v rámci klinických studií. Jde o malé molekuly, které jsou schopny vazby na defektní enzym v endoplazmatickém retikulu. Tím modifikují jeho terciární strukturu a umožňují transport enzymu do lysosomu, kde může uplatnit svoji enzymatickou aktivitu. Vazba chaperonu je reverzibilní, předpokladem je zachovaná částečná tvorba vlastního enzymu.
Další možnosti léčby
Další možnosti léčby jako transplantace kostní dřeně, kmenových buněk či genová terapie nejsou u Gaucherovy nemoci využívány pro nedostatečný účinek a množství závažných nežádoucích účinků. Splenektomie byla dříve často indikována u pacientů s těžkou trombocytopenií, masivní splenomegalií s projevy hypersplenismu a krvácivé diatézy. Častou komplikací u pacientů po splenektomii je nezvládnutelná sepse či zhoršení skeletálního postižení při masivním střádání. V současné době je možné významně ovlivnit celkový stav pacienta pomocí enzymatické substituční léčby a splenektomie již není kromě výjimečných stavů doporučována.
Vzhledem k multisystémovému postižení a k progresivnímu charakteru nemoci je nutno pacientům poskytnout komplexní péči včetně sociální a symptomatické léčby. Nezbytné je genetické poradenství v rodinách pacientů s Gaucherovou nemocí včetně vyšetření partnerů a zajištění prenatální diagnostiky.
Kasuistika 1
Pacient ve věku 24 let, narodil se z první fyziologické gravidity s porodní hmotností 4 030 g a s porodní délkou 51 cm. Poporodní adaptace i psychomotorický vývoj byly v normě. Od tří let byl dispenzarizován pro hepatosplenomegalii 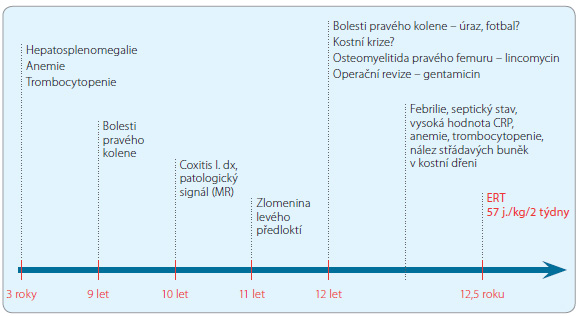 a mírnou anemii. Ve věku devíti let se nejdříve po fyzické zátěži a později i trvale objevily bolesti pravého kolenního kloubu. Vyšetření MR ukázalo patologický signál dřeně v distální polovině pravého femuru bez periostální reakce, nález byl hodnocen jako pravděpodobný následek posttraumatických změn. Ve věku 10 let byl chlapec pro akutně vzniklé bolesti pravého kyčelního kloubu léčen antibiotiky pro klinickou diagnózu coxitis l. dx., obtíže ustoupily, ve stejném období byla dle sérologie diagnostikována borrelióza, po antibiotické léčbě se sérologické nálezy upravily. V 11 letech si při sportu zlomil levé předloktí, fraktura se zhojila bez komplikací. Krátce poté se objevil otok a bolestivost pravého kolenního kloubu s alterací celkového stavu a s vysokými teplotami. Pro podezření na akutní osteomyelitidu byl na ortopedii léčen lincomycinem, byla provedena operační revize s implantací gentamicinu. Klinicky se stav přechodně upravil, ale po třech týdnech se znovu objevily septické teploty, progredovala splenomegalie, anemie, leukopenie, trombocytopenie a koagulopatie s prodlouženým APTT a s vysokými hodnotami D‑dimerů (obr. 11, tab. 1). Pro podezření na hemoblastózu byla vyšetřena kostní dřeň s nálezem pěnovitých buněk svědčících pro Gaucherovu nemoc.
a mírnou anemii. Ve věku devíti let se nejdříve po fyzické zátěži a později i trvale objevily bolesti pravého kolenního kloubu. Vyšetření MR ukázalo patologický signál dřeně v distální polovině pravého femuru bez periostální reakce, nález byl hodnocen jako pravděpodobný následek posttraumatických změn. Ve věku 10 let byl chlapec pro akutně vzniklé bolesti pravého kyčelního kloubu léčen antibiotiky pro klinickou diagnózu coxitis l. dx., obtíže ustoupily, ve stejném období byla dle sérologie diagnostikována borrelióza, po antibiotické léčbě se sérologické nálezy upravily. V 11 letech si při sportu zlomil levé předloktí, fraktura se zhojila bez komplikací. Krátce poté se objevil otok a bolestivost pravého kolenního kloubu s alterací celkového stavu a s vysokými teplotami. Pro podezření na akutní osteomyelitidu byl na ortopedii léčen lincomycinem, byla provedena operační revize s implantací gentamicinu. Klinicky se stav přechodně upravil, ale po třech týdnech se znovu objevily septické teploty, progredovala splenomegalie, anemie, leukopenie, trombocytopenie a koagulopatie s prodlouženým APTT a s vysokými hodnotami D‑dimerů (obr. 11, tab. 1). Pro podezření na hemoblastózu byla vyšetřena kostní dřeň s nálezem pěnovitých buněk svědčících pro Gaucherovu nemoc.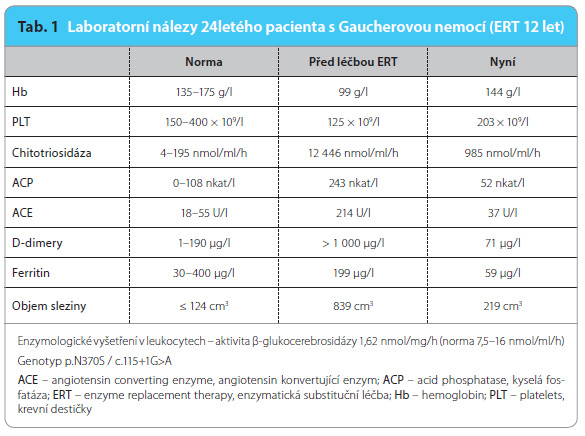
Při metabolickém vyšetření byla nápadná bledost kůže a sliznic, mírná tachykardie a omezená hybnost pravé dolní končetiny po operační revizi. Chlapec měl výraznou hepatosplenomegalii – játra zasahovala 5 cm pod oblouk žeberní, slezina dosahovala k pupku. Mentální úroveň odpovídala věku.
V laboratorním vyšetření byla zjištěna vysoká aktivita kyselé fosfatázy a chitotriosidázy svědčící pro aktivaci makrofágového systému a zvýšená koncentrace angiotensin konvertujícího enzymu. Vyšetření MR zobrazilo infiltraci obratlových těl Th, L, S páteře i dřeně kostí dlouhých končetin. Denzitometrické vyšetření ukázalo snížený obsah kostního minerálu v bederní páteři (Z‑skóre –2,3). Volumometrické vyšetření jater a sleziny pomocí MR potvrdilo hepatomegalii (objem 1 419 cm3, norma pro věk ≤ 800 cm3) a splenomegalii (839 cm3, norma < 80 cm3). Diagnózu Gaucherovy nemoci verifikovalo enzymologické vyšetření β‑glukocerebrosidázy v izolovaných leukocytech (1,62 nmol/ml/h, norma 7,5–16 nmol/ml/h) a DNA vyšetření genu pro β‑glukocerebrosidázu. Chlapec je složený heterozygot pro p.N370S a sestřihovou mutaci c.115+1G>A, která na úrovni transkriptu vede k vypuštění celého exonu 2. Rodiče jsou přenašeči těchto mutací.
Po zhodnocení klinického stavu byla zahájena enzymatická substituční léčba rekombinantní β‑glukocerebrosidázou (imigluceráza – 60 j./kg/2 týdny, postupně 30 j./kg/2 týdny). Do půl roku vymizely bolesti kloubů a kostí a upravily se hematologické parametry. Po roce léčby dochází k ústupu organomegalie. Kontrolní vyšetření MR po dvou letech dokumentuje ústup infiltrace kostní dřeně obratlů i dlouhých kostí, přetrvává pouze mírná elevace chitotriosidázy.
Kasuistika 2
Pacientka ve věku 64 let, do věku tří let vážněji nestonala, prospívala dobře, psychomotorický vývoj byl fyziologický. Ve třech letech prodělala spálu, poté se objevilo nechutenství, nízké hmotnostní přírůstky, opožďování růstu. Od šesti let udávala bolesti dlouhých kostí dolních končetin, které byly vyhodnoceny jako růstové bolesti. V šesti letech byla provedena adenotomie a tonsilektomie, v deseti letech znovu adenotomie. V průběhu předoperačního vyšetření byla zjištěna splenomegalie a trombocytopenie, v punktátu kostní dřeně byly nalezeny střádavé buňky. Podezření na Gaucherovu chorobu bylo potvrzeno enzymologickým vyšetřením se zjištěnou sníženou aktivitou β‑glukocerebrosidázy v leukocytech (1,27 nmol/mg/h, norma 7,5–16 nmol/mg/h) a s nálezem specifické mutace (genotyp p.N370S/p.R163*). Pro zhoršující se trombocytopenii byla indikována splenektomie ve věku deseti let. Po odstranění sleziny sice vzrostl počet trombocytů, intenzivní střádání však pokračovalo v dalších lokalizacích.
V důsledku oslabení kortikální kosti a předčasné osteoporózy prodělala několik patologických fraktur – v 15 letech frakturu levého bérce s následnou osteosyntézou, ve 49 letech frakturu pravého femuru s nutností osteosyntézy hřebem a reosteosyntézy kondylární dlahou s následnou spongioplastikou pro špatné hojení, v 55 letech tříštivou zlomeninu distální metafýzy pravého femuru s následným operačním řešením osteosyntézou. Pooperační stav byl opakovaně komplikován flebotrombózou pravé dolní končetiny s nutností přechodné warfarinizace. Ve 49 letech prodělala kostní krizi provázenou krutými bolestmi kostí, imobilizací, teplotou a závažnou anemií, pro kterou byl nutný opakovaný převod erymasy. V 51 letech byla provedena totální endoprotéza kyčelního kloubu pro těžké artrotické změny hlavice pravého femuru.
Pacientka prodělala další závažná infekční onemocnění septického charakteru. Ve 47 letech byla hospitalizována pro septické teploty a susp. plicní embolii, kultivačně byla prokázána salmonelová etiologie obtíží. Ve 48 letech byl stav komplikován závažně probíhající bakteriální endokarditidou a akutní pyelonefritidou charakteru urosepse. Ve věku 60 let byla pacientce indikována cholecystektomie pro cholecystolitiázu a subjektivní obtíže.
Závažné obtíže byly zaznamenány i v gynekologické anamnéze – ve 20 letech bylo indikováno ukončení těhotenství ze zdravotních důvodů, ve 23 letech porodila zdravého syna. Dále prodělala spontánní potrat ve věku 26 a 29 let. Ve 32 letech byla sledována pro rizikové těhotenství zakončené v termínu spontánním porodem zdravého syna. V důsledku peripartálního krvácení byly opakovaně nutné transfuze erymasy.
Enzymatická substituční léčba imiglucerázou byla zahájena až ve věku 52 let (obr. 12, tab. 2). V té době již byla v důsledku osteoporózy fixována řada ireverzibilních změn. Přesto jsme zhruba šest měsíců po zahájení léčby pozorovali ústup subjektivních obtíží, zmenšila se únava, zmírnily se bolesti kostí a kloubů, upravily se hematologické parametry. V důsledku splenektomie nebyla před léčbou v popředí obtíží významnější trombocytopenie, spíše mírná anemie. Rizikem splenektomie jsou však septické komplikace mnohdy s fatálním průběhem a vystupňované postižení skeletu. Postupně došlo k poklesu koncentrace biomarkerů, k ústupu hepatomegalie, na MR je zjevná zástava progrese skeletálního postižení. V průběhu enzymoterapie a při současném podávání bisfosfonátů, vápníku a vitaminu D verifikujeme denzitometricky mírné zlepšení osteoporózy v oblasti lumbální páteře, levé kyčle a levého předloktí.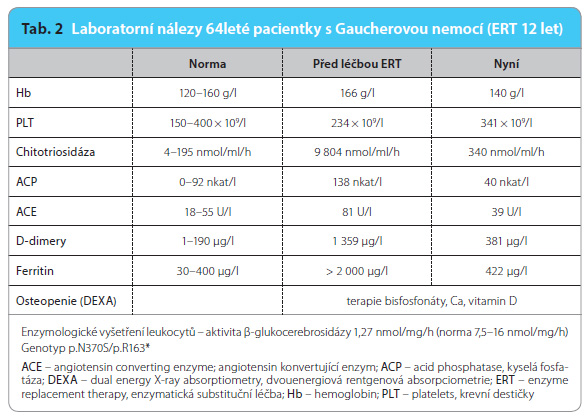
Je zjevné, že při včasném zahájení léčby by nebylo nutno indikovat splenektomii a ohrozit pacientku septickými komplikacemi. Rovněž lze předpokládat, že by nedošlo k tak závažnému skeletálnímu postižení s opakovanými patologickými frakturami a s nutností ortopedických korekcí. Předčasný rozvoj osteoporózy, těžkých kloubních artrotických změn, trombotických i gynekologických komplikací je typický pro středně závažný průběh neléčené Gaucherovy nemoci. Je proto nezbytné stanovit diagnózu co nejdříve a zahájit enzymatickou substituční léčbu při zjevné progresi onemocnění před rozvojem ireverzibilního poškození tkání či orgánů.
Situace v České republice
V České republice je v současné době sledováno 35 pacientů s prokázanou diagnózou Gaucherovy nemoci, z nich jeden pacient trpí III. typem, ostatní I. typem. Ve sledovaném souboru pacientů je 12 mužů a 23 žen, průměrný věk pacientů je 38 let (4,5–64 let). Průměrný věk, kdy byly zaznamenány první příznaky Gaucherovy nemoci, činí 4,5 roku s rozmezím 0,5–49 let. Diagnóza Gaucherovy nemoci byla stanovena průměrně ve 22 letech (1,5–49 let), období od prvních příznaků do stanovení diagnózy odpovídá přibližně 6,7 roku (1 měsíc až 25 let). Doba od stanovení diagnózy Gaucherovy nemoci do zahájení specifické léčby je přibližně 5,8 roku (2 měsíce až 22 let), od prvních příznaků do zahájení léčby pak 12 let (2,5 měsíce až 43 let). Do posledních dvou údajů nejsou zahrnuti pacienti, kteří jsou asymptomatičtí či mají minimální obtíže, jsou pouze sledováni, ale nejsou léčeni. Jde většinou o sourozence pacientů s touto nemocí, u nichž byla diagnóza Gaucherovy nemoci stanovena v rámci vyšetření v rodině. Enzymatickou substituční terapií je léčeno 28 pacientů (21 pacientů je léčeno imiglucerázou, 7 velaglucerázou), substrát redukující terapie je podávána jednomu pacientovi, 6 pacientů není léčeno. Dle naší zkušenosti je účinek obou přípravků používaných v ČR k enzymatické substituční terapii srovnatelný. Všichni pacienti jsou sledováni v našem Centru pro léčbu střádavých onemocnění na Klinice dětského a dorostového lékařství 1. LF UK a VFN v Praze.
Závěr
Gaucherova nemoc je jedním z nejčastějších střádavých onemocnění, které má bez léčby trvale progresivní charakter s ovlivněním kvality života a zkrácením doby dožití. Je nutno stanovit diagnózu co nejdříve a u symptomatických pacientů zahájit včas vhodnou léčbu. Jen tak lze předejít rozvoji ireverzibilního poškození včetně skeletálních změn, kloubního postižení, rozvoje osteoporózy, hypersplenismu, cirhózy jater či portální hypertenze s následnými krvácivými komplikacemi. Jde o multiorgánové a multisystémové postižení, pacienty je proto vhodné sledovat v centrech, kde je zajištěna komplexní péče včetně ambulantních specialistů, kteří mají zkušenost s projevy a možnými komplikacemi střádavých onemocnění. Nezbytnou součástí péče je sociální a genetické poradenství v rodinách pacientů včetně plánovaného rodičovství a prenatální diagnostiky.
Práce byla podpořena grantem MZ ČR – RVO VFN 64165.
Seznam použité literatury
- [1] Poupetova H, Ledvinova J, Berna L, et al. The birth prevalence of lysosomal storage disorders in the Czech Republic: comparison with data in different populations. J Inherit Metab Dis 2010; 33: 387–396.
- [2] Vellodi A. Lysosomal storage disorders. Br J Haematol 2005; 128: 413–431.
- [3] Conradi NG, Sourander P, Nilsson O, et al. Neuropathology of the Norrbottnian type of Gaucher disease. Morphological and biochemical studies. Acta Neuropathol 1984; 65: 99–109.
- [4] Svennerholm L, Erikson A, Groth CG, et al. Norrbottnian type of Gaucher disease – clinical, biochemical and molecular biology aspects: successful treatment with bone marrow transplantation. Dev Neurosci 1991; 13: 345–351.
- [5] Malinová V, Honzík T. Lysosomální onemocnění – současné možnosti diagnostiky a terapie. Pediatr praxi 2013; 14: 99–104.
- [6] Vitner EB, Farfel Becker T, Eilam R, et al. Contribution of brain inflammation to neuronal cell death in neuronopathic forms of Gaucher‘s disease. Brain 2012; 135(Pt 6): 1724–1735.
- [7] Allen MJ, Myer BJ, Khokher AM, et al. Pro inflammatory cytokines and the pathogenesis of Gaucher´s disease: Increased release of interleukin 6 and interleukin 10. QJM 1997; 90: 19–25.
- [8] Michelakakis H, Spanou C, Kondyli A, et al. Plasma tumor necrosis factor α (TNF α) levels in Gaucher disease. Biochim Biophys Acta 1996; 1317: 219–222.
- [9] de Fost M, Out TA, de Wilde FA, et al. Immunoglobulin and free light chain abnormalities in Gaucher disease type I: data from an adult cohort of 63 patients and review of the literature. Ann Hematol 2008; 87: 439–449.
- [10] Gaucher disease and Cerezyme® monograph – ©2005 Genzyme Corporation.
- [11] Wenstrup RJ, Roca Espiau M, Weinreb NJ, Bembi B. Skeletal aspects of Gaucher disease: a review. Br J Radiol 2002; 75 Suppl 1: A2–12.
- [12] Granovsky Grisaru S, Belmatoug N, vom Dahl S, et al. The management of pregnancy in Gaucher disease. Eur J Obstet Gynecol Reprod Biol 2011; 156: 3–8.
- [13] Lwin A, Orvisky E, Goker Alpan O, et al. Glucocerebrosidase mutations in subjects with parkinsonism. Mol Genet Metab 2004; 81: 70–73.
- [14] Mitsui J, Mizuta I, Toyoda A, et al. Mutations for Gaucher disease confer high susceptibility to Parkinson disease. Arch Neurol 2009; 66: 571–576.
- [15] Sidransky E, Nalls MA, Aasly JO, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson‘s disease. N Engl J Med 2009; 361: 1651–1661.
- [16] Westbroek W, Gustafson AM, Sidransky E. Exploring the link between glucocerebrosidase mutations and parkinsonism. Trends Mol Med 2011; 17: 485–493.
- [17] Lo SM, Stein P, Mullaly S, et al. Expanding spectrum of the association between Type 1 Gaucher disease and cancers: a series of patients with up to 3 sequential cancers of multiple types – correlation with genotype and phenotype. Am J Hematol 2010; 85: 340–345.
- [18] Zimran A, Altarescu G, Rudensky B, et al. Survey of hematological aspects of Gaucher disease. Hematology 2005; 10: 151–156.
- [19] Tayebi N, Stone DL, Sidransky E. Type 2 Gaucher disease: an expanding phenotype. Mol Genet Metab 1999; 68: 209–219.
- [20] Abrahamov A, Elstein D, Gross Tsur V, et al. Gaucher´s disease variant characterised by progressive calcification of heart valves and unique genotype. Lancet 1995; 346: 1000–1003.
- [21] Green BA, Alexander AA, Hill PR, et al. Imaging findings in pediatric type 1 Gaucher disease: what the clinician needs to know. J Pediatr Hematol Oncol 2011; 33: 301–305.
- [22] Itzchaki M, Lebel E, Dweck A, et al. Orthopedic considerations in Gaucher disease since the advent of enzyme replacement therapy. Acta Orthop Scand 2004; 75: 641–653.
- [23] Saudubray JM, et al. Inborn Metabolic Diseases: diagnosis and treatment. 5th edition; Berlin Heidelberg: Springer, 2012.
- [24] Hodanova K, Hrebicek M, Cervenkova M, et al. Analysis of the beta glukocerebrosidase gene in Czech and Slovak Gaucher patients: mutation profile and description of six novel mutant alleles. Blood Cells Mol Dis 1999; 25: 287–298.
- [25] Hruska KS, LaMarca ME, Scott CR, et al. Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum Mutat 2008; 29: 567–583.
- [26] Koprivica V, Stone DL, Park JK, et al. Analysis and classification of 304 mutant alleles in patients with type 1 and type 3 Gaucher disease. Am J Hum Genet 2000; 66: 1777–1786.
- [27] Grabowski GA. Gaucher Disease. Enzymology, genetics, and treatment. Adv Hum Genet 1993; 21: 377–441.
- [28] Zimran A, Altarescu G, Phillips M, et al. Phase 1/2 and extension study of velaglucerase alfa replacement therapy in adults with type 1 Gaucher disease: 48 month experience. Blood 2010; 115: 4651–4656.
- [29] SUKL http://www.sukl.cz‚ Souhrn údajů o přípravku – Cromiva®, revize textu z 10. 12. 2014.
- [30] Harrison L. Evidence Mounting for Eliglustat in Gaucher‘s Disease. Medscape Medical News. http://www.medscape.com/viewarticle/834451. Navštíveno: November 10, 2014.
- [31] European Medicines Agency; EU summary of product characteristics: Cerdelga®. http://www.ema.europa.eu/docs/cs_CZ/document_library/EPAR_ _Product_Information/human/003724/WC500182387.pdf Navštíveno: 6. 3. 2016