Hepatotoxicita inhibitorů kináz
Souhrn:
Pavlík T. Hepatotoxicita inhibitorů kináz. Remedia 2019; 29: 284–290.
Léky indukované poškození jater je jednou z častých komplikací léčby. Dokonce je uváděno, že až 50 % perorálně užívaných léčiv je spo-jeno s tímto nežádoucím účinkem. Ten může být jedním z rozhodujících kritérií pro omezení používání léčiva či pro stažení z trhu. Novou skupinou léčiv, která je také spojena s hepatotoxicitou, jsou inhibitory kináz. Dosud převažuje jejich využití v onkologii, nicméně tato cílená terapeutická strategie si nachází stále významnější roli v léčbě dalších onemocnění, jako jsou revmatoidní artritida, idiopatická plicní fibróza, idiopatické střevní záněty. Že si tato skupina léčiv zasluhuje zvýšenou pozornost, dokládá skutečnost, že u řady z nich pří-balová informace obsahuje tzv. varování v rámečku, které se týká právě hepatotoxicity.
Summary:
Pavlik T. Hepatotoxicity of kinase inhibitors. Remedia 2019; 29: 284–290.
One the common treatment complications are drug induced liver injury. It is even reported that as much as 50% of oral drugs is associ-ated with this adverse effect. It may be one of the criteria for the decision to limit administration of the drug or to withdraw the drug from the market. Kinase inhibitors are a new drug category associated with hepatotoxicity. So far, their use is limited to oncology. However, the role of this targeted therapeutical strategy is becoming more important in the treatment of other diseases, for example rheumatoid arthritis, idiopatic pulmonary fibrosis and idiopatic inflammatory bowel disease. The fact that this drug category requires increased caution is evidenced by a so called box warning reagrding hepatotoxicity contained in the patient information leaflet.
Key words: kinase inhibitors, hepatotoxicity, DILI.
Polékové poškození jater patří obecně mezi časté komplikace
terapie. V literatuře je uváděno, že více než 900 léčivých přípravků má
hepatotoxický potenciál [1]. Z rozsáhlé srovnávací studie z roku 2015
dokonce vyplývá, že téměř 50 %
registrovaných léčiv má potenciál indukovat jaterní poškození různého stupně
[2]. Toto se samozřejmě netýká pouze léčiv. Poškození jater mohou vyvolat také
potravní doplňky či rostlinné přípravky [3]. Význam problematiky léčivy
indukovaného jaterního poškození (DILI, drug‑induced liver injury) je
na vzestupu, především pak s ohledem na stále rostoucí počet
léčiv. Intenzita jaterního poškození je velmi široká, a to od mírného
zvýšení aktivity jaterních enzymů (ALT, AST, GGT) či hodnoty bilirubinu až
po těžké stavy s infaustní prognózou. Při hodnocení pacienta
s nově diagnostikovaným poškozením jater by vždy měla být farmakoterapie
považována za možnou příčinu, a to i tehdy, kdy je zřejmá jiná
příčina, jako je hepatitida B nebo C [4]. Důvod je nasnadě, při ukončení
podávání hepatotoxického léčiva obvykle dochází k úpravě stavu. Rozpoznání
případu, kdy je léčivo příčinou jaterního poškození, je nicméně obtížné.
Klinický obraz je často variabilní a může napodobovat téměř jakoukoliv
formu onemocnění jater – např. akutní hepatitidu, chronickou hepatitidu,
akutní selhání jater, obstrukci žlučových cest nebo jaterní steatózu.
DILI – léky indukované poškození jater
Léky indukované poškození jater patří mezi jeden
z nejvýznamnějších nežádoucích účinků léčiv. Pokud se jedná
o predikovatelné poškození jater, a to na základě podané dávky
či přímých farmakotoxických účinků léčiva, hovoříme o tzv. vnitřním typu
(typicky např. předávkování paracetamolem). Druhým typem je idiosynkratické
poškození jater, které lze jen obtížně predikovat. Předpokládá se, že se jedná
o komplexní stav, který je determinován jak vlastnostmi léčiva, tak stavem
pacienta samotného [5].
Z diagnostického hlediska je vhodné rozdělení
hepatotoxicity na typ hepatocelulární, cholestatický nebo smíšený. Dělení
vychází z histologických charakteristik poškození a míry elevace
hodnot jaterních enzymů. Z diagnostického hlediska má význam rovněž
klasifikace na základě tzv. klinického fenotypu, ta je založena
na podobnosti s jinými jaterními onemocněními, jako je např. akutní
hepatitida, cholangitida, autoimunitní hepatitida, cirhóza apod.
Hepatocelulární typ
Jedná se o léčivem indukované poškození jater, které se
podobá akutní virové hepatitidě. Biopsie jater obvykle vykazuje výraznou
nekrózu a zánět hepatocytů. Stáze žluči je spíše mírná, tedy alespoň
v počátečních stadiích. Z celkových příznaků převažuje únava a slabost.
Hodnoty alaninaminotransferázy (ALT) a aspartátaminotransferázy (AST) jsou
typicky značně zvýšené (obvykle > 10krát), zatímco alkalická
fosfatáza a gamaglutamyltransferáza (GGT) mají hodnoty zvýšeny jen mírně.
Typicky je poměr aktivity ALT k hodnotě alkalické fosfatázy (poměr „R“,
vyjádřen jako násobky horní hranice jejich normálních hodnot) ≥ 5
[4]. Mezi látky, které typicky vykazují hepatocelulární poškození, patří
například paracetamol, isoniazid, nitrofurantoin a metyldopa, statiny
a další [1,4].
Cholestatický typ
Cholestatický typ poškození jater obvykle připomíná
obstrukci žlučovodů nebo choledocholitiázu. Z celkových projevů převládá
žloutenka a pruritus. Rovněž může být přítomna bolest v pravém horním
kvadrantu. Aktivita alkalické fosfatázy stejně jako hodnoty GGT
a bilirubinu jsou výrazně zvýšené, naopak aktivity ALT a AST jsou
zvýšeny minimálně nebo jen mírně. Poměr ALT k alkalické fosfatáze je
obvykle ≤ 2. Mezi léky, které zpravidla způsobují cholestatické
poškození jater, patří například amoxicilin/klavulanát, steroidy, tricyklická
antidepresiva, ciprofloxacin, sulfonylmočovina a další [1,4].
Smíšený (hepatocelulárně‑cholestatický) typ
Jedná se o jaterní poškození, které je „typické“ pro
léčiva, neboť se jen zřídka vyskytuje u jiných forem akutního jaterního
onemocnění. Jaterní biopsie vykazuje prominentní nekrózu a zánět
hepatocytů, který je doprovázen výraznou stazí žluči. Symptomy mohou zahrnovat
jak únavu, tak svědění. Laboratorní testy ukazují zvýšení aktivity ALT
a alkalické fosfatázy v séru. Poměr ALT k alkalické fosfatáze se
pohybuje v rozmezí 2−5. Mezi léčiva, která mohou být příčinou smíšeného
typu jaterního poškození, patří například sulfonamidy, ibuprofen, fenytoin,
enalapril a řada dalších [1,4].
Hepatotoxicita inhibitorů kináz
Inhibitory kináz (KI) jsou dnes početnou skupinou léčiv, jež
specificky cílí na proteinové kinázy. Ty jsou umístěny buďto
na vnitřní straně buněčné membrány, nebo intracelulárně. Hrají rozhodující
roli v buněčné signalizaci, která se podílí na metabolismu, obranných
reakcích, adaptaci, růstu a na diferenciaci buněk. Podkladem aktivace
kináz je přenesení fosfátové skupiny z ATP (fosforylace)
na specifický substrát, obvykle na aminokyselinu tyrozin, a též na
serin a threonin.
Lidský genom obsahuje informace kódující více než 500 proteinových
kináz. Jak bylo uvedeno, proteinové kinázy mohou být specificky zapojeny
do buněčného růstu, proliferace a diferenciace. Z toho plyne, že
mutace genů kódujících kinázy mohou vést
k neregulovanému růstu a proliferaci, která je typická především pro
nádorové a imunitní buňky. I proto zmíněné mutace proteinových kináz
představují cíl (cílená terapie) pro řadu protinádorových léčiv. Oblasti
využití KI se nicméně neustále rozšiřují. Již dnes jsou využívány například
v revmatologii (revmatoidní artritida − tofacitinib),
gastroenterologii (idiopatické střevní záněty − tofacitinib,
filgotinib) a v pneumologii (idiopatická plicní
fibróza − nintedanib).
Zavedení KI do klinické praxe zásadním způsobem
ovlivnilo léčebné strategie uvedených oborů. Přes nesporný přínos bylo
zjištěno, že jejich použití je spojeno s rizikem vzniku závažných
nežádoucích účinků, včetně hepatotoxicity. Ta se týká většiny dosud schválených
KI. V případě lapatinibu, pazopanibu, ponatinibu, regorafenibu
a sunitinibu jsou důkazy natolik silné a závažné, že součástí
příbalové informace léčivých přípravků registrovaných prostřednictvím
amerického Úřadu pro kontrolu potravin a léčiv (FDA) je také tzv. varování
v rámečku (black‑box warnings) [6].
Nástup hepatotoxicity je obvykle pozorován v průběhu
prvních dvou měsíců (2−8 týdnů) od zahájení léčby, může být ale
i opožděn (tab. 1). Ačkoliv se zpravidla jedná o vratný
proces, léčba pomocí KI může v některých případech vést k trvalému
poškození jater. Toxicita je zpravidla zvládnutelná úpravou dávky, přechodem
na alternativní KI s nižším hepatotoxickým potenciálem, eventuálně
úplným 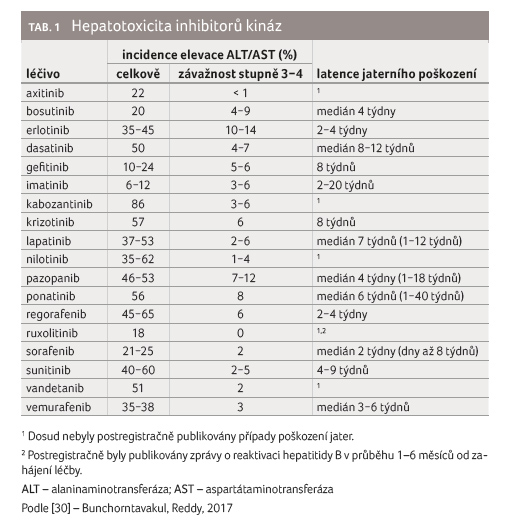 ukončením léčby. Dle očekávání se jedná zpravidla o toxicitu
hepatocelulárního typu, nicméně byly popsány i vzácné případy cholestázy
[6]. Projevy hypersenzitivity obvykle nejsou přítomny. Zajímavá spojitost mezi
příčinou hepatotoxicity a imunitním systémem HLA je přesto pozorována
u lapatinibu, kdy bylo prokázáno četnější zastoupení variantní alely
DQA1*02:01 u pacientů léčených lapatinibem a současně se zvýšenými
aktivitami aminotransferáz ve srovnání s pacienty s běžnými
aktivitami aminotransferáz (71 %
vs. 21 %) [7]. Úmrtí
vzniklá v přímé souvislosti s léčbou KI jsou velmi vzácná. Popsána
jsou například u krizotinibu, erlotinibu, imatinibu, lapatinibu, pazopanibu,
ponatinibu, regorafenibu a sunitinibu [8−13]. Pacienti by měli být proto
pod důkladnou kontrolou, což potvrzuje řada postmarketingových studií [4,6].
ukončením léčby. Dle očekávání se jedná zpravidla o toxicitu
hepatocelulárního typu, nicméně byly popsány i vzácné případy cholestázy
[6]. Projevy hypersenzitivity obvykle nejsou přítomny. Zajímavá spojitost mezi
příčinou hepatotoxicity a imunitním systémem HLA je přesto pozorována
u lapatinibu, kdy bylo prokázáno četnější zastoupení variantní alely
DQA1*02:01 u pacientů léčených lapatinibem a současně se zvýšenými
aktivitami aminotransferáz ve srovnání s pacienty s běžnými
aktivitami aminotransferáz (71 %
vs. 21 %) [7]. Úmrtí
vzniklá v přímé souvislosti s léčbou KI jsou velmi vzácná. Popsána
jsou například u krizotinibu, erlotinibu, imatinibu, lapatinibu, pazopanibu,
ponatinibu, regorafenibu a sunitinibu [8−13]. Pacienti by měli být proto
pod důkladnou kontrolou, což potvrzuje řada postmarketingových studií [4,6].
Potvrzení poškození jater v důsledku léčby KI je
nicméně komplikované. Významnou roli zde hraje řada faktorů, v neposlední
řadě přítomnost jaterních metastáz, současně užívaná léčiva či lékové interakce
[6]. Spolehlivost
predikce DILI také komplikuje dosud omezená znalost specifických biomarkerů
[5].
Příčiny hepatotoxicity inhibitorů kináz
Mechanismus vzniku
hepatatoxicity v důsledku léčby KI není dosud zcela objasněn. V této
souvislosti je proto velmi přínosná studie z roku 2018, která zkoumala
mechanismy hepatotoxicity u šesti KI (regorafenibu, sorafenibu,
ponatinibu, krizotinibu, dasatinibu a pazopanibu) za využití lidské
nádorové buněčné linie HepG2 a hepatomové linie HepaRG [14]. Ta prokázala,
že regorafenib a sorafenib silně inhibují oxidační metabolismus
a glykolýzu, snižují potenciál mitochondriální membrány a indukují
apoptózu a/nebo nekrózu buněk HepG2 v koncentracích
podobných plazmatickým koncentracím rovnovážného stavu u lidí.
U těchto látek se zdá, že toxicita je spojena především se samotnou
účinnou látkou a méně pak s metabolity. Rovněž v případě
ponatinibu byla popsána inhibice oxidačního metabolismu. Glykolýza byla naopak
ovlivněna méně významně. Oproti regorafenibu a sorafenibu však docházelo
k apoptóze buněk HepG2 v koncentracích
vyšších, než jsou plazmatické koncentrace v rovnovážném stavu u lidí.
Naopak krizotinib a dasatinib, ačkoliv významně neovlivňovaly
mitochondriální funkce a inhibovaly glykolýzu pouze slabě, apoptózu buněk
HepG2 indukovaly i při koncentracích podobných plazmatickým. Při léčbě
pazopanibem bylo prokázáno mírné zvýšení koncentrací mitochondriálních
reaktivních forem kyslíku a inhibice glykolýzy. Z výše uvedeného tedy
vyplývá, že mitochondriální toxicita a inhibice glykolýzy s největší
pravděpodobností vysvětlují hepatotoxicitu spojenou s regorafenibem,
sorafenibem a případně s pazopanibem. Pro ostatní molekuly je potřeba
dalšího výzkumu [14].
Hepatotoxicitou KI se rovněž zabývala studie z roku
2017 [15]. Zkoušeno bylo celkem 31 molekul, což odpovídalo v té době KI
aktuálně registrovaným prostřednictvím FDA. Oproti výše zmíněné studii
z roku 2018 byly zkoušky prováděny na mitochondriích izolovaných
z jater potkanů. Testovány byly koncentrace KI v rozmezí
od terapeutických maximálních koncentrací v krvi (cmax)
až po 100násobné hodnoty cmax. Pozornost byla
soustředěna na celou řadu parametrů, např. na spotřebu kyslíku,
potenciál na vnitřní straně membrány, uvolnění cytochromu C
z mitochondrie, reaktivní formy kyslíku a na individuální
aktivity jednotlivých komplexů dýchacího řetězce (I–V). Z 31 vyšetřovaných
KI byly pouze tři spojeny s významnou mitochondriální toxicitou při
koncentracích rovných cmax (sorafenib, regorafenib,
pazopanib). To potvrzuje závěry výše zmíněné studie, že mitochondriální
toxicita je v přímém spojení s patogenezí hepatotoxicity tří
zmíněných KI [15]. Při sledování koncentrací rovných 100násobku cmax bylo indukováno mitochondriální poškození pouze u 18
KI. Určitým překvapením bylo, že z látek patřících do skupiny
s povinným varováním o hepatotoxicitě, tzv. black‑box warnings, bylo
indukováno mitochondriální poškození pouze u regorafenibu, lapatinibu,
idelalisibu a pazopanibu, ale nikoliv u ponatinibu a sunitinibu
[15].
Ačkoliv tedy uvedená data ukazují na to, že
mitochondriální toxicita může pomoci vysvětlit hepatotoxicitu KI či
identifikovat potenciálně hepatotoxické KI, existují nepochybně další
mechanismy a/nebo rizikové faktory, které mohou s hepatoxicitou KI
souviset. Ty mohou zahrnovat metabolickou aktivaci [16,17], přímé
mitochondriální poškození [18], oxidační stres [19], inhibici jaterních transportních
mechanismů [20] nebo geneticky podmíněnou variabilitu enzymů metabolizujících
KI [21,22].
Další doplnění znalostí mechanismů vzniku jaterního
poškození v důsledku léčby KI přinesly závěry studie z roku 2017
[23]. Ta se soustředila na hepatotoxicitu imatinibu, sunitinibu,
lapatinibu a erlotinibu. Opět byly využity buněčné linie HepG2, HepaRG
a mitochondrie myších jaterních buněk. Studie prokázala, že imatinib,
sunitinib a méně lapatinib jsou spojeny s mitochondriální dysfunkcí a inhibicí
glykolýzy v koncentracích dosahovaných v hepatocytech léčených
pacientů. Současně indukce cytochromu P450 pomocí rifampicinu zvýšila toxicitu,
což naznačuje tvorbu toxických metabolitů. V případě erlotinibu nebyla
prokázána souvislost s mitochondriální dysfunkcí ani s poruchou
glykolýzy, dokonce indukce cytochromu P450 vedla ke snížení
hepatocelulární toxicity. To naznačuje, že poškození jater je spojeno především
s mateřskou látkou [23].
V literatuře jsou dále popsány případy, kdy poškození
jater po podání KI vykazovalo znaky autoimunitních procesů [4]. Podání
imatinibu a nilotinibu bylo rovněž spojováno s reaktivací hepatitidy
B. Nebylo nicméně zřejmé, zda se toto týká specifické aktivity uvedených KI
na replikaci viru hepatitidy typu B, nebo zda jde o důsledek
imunosuprese [24].
LIVERTOX jako zdroj informací
Jak bylo uvedeno v úvodu článku, hepatotoxicita jako
nežádoucí účinek léčiv je poměrně častá. Přesto bývá složité vyhledat dostupné
a přehledné informace k této problematice. I to bylo důvodem,
proč americký Národní institut pro diabetes, onemocnění trávicí soustavy
a ledvin (NIDDK, The National Institute of Diabetes and Digestive and
Kidney Diseases), spadající pod National Institutes of Health (NIH),
ve spolupráci s Národní lékařskou knihovnou (NLM) vytvořil veřejně
a volně dostupný zdroj informací – databázi LIVERTOX (www.livertox.org). Ta je zaměřena
na poskytování aktuálních a komplexních informací ve vztahu
k problematice DILI. Cílem projektu je obsáhnout nejen všechna dostupná
léčiva, ale také doplňky stravy. Charakteristika léčiv zahrnuje chemickou
strukturu, indikace, doporučené dávky, především pak stručný popis klinického
obrazu a průběhu jaterního poškození. V klinické praxi pak může být
pro rozhodování rovněž přínosné tzv. skóre pravděpodobnosti [4].
Skóre pravděpodobnosti vzniku jaterního poškození
Inhibitory kináz lze členit podle míry pravděpodobnosti
vzniku jaterního poškození do sedmi kategorií. Každá kategorie je pak
spojena s určitou mírou pravděpodobnosti vzniku jaterního poškození –
skóre pravděpodobnosti (tab. 2). Kategorizace vychází především
z veřejně dostupných literárních zdrojů. Čím déle je léčivo
na trhu, tím je spolehlivost kategorizace vyšší. Prostřednictvím sítě
DILIN (Drug‑Induced Liver Injury Network) je klasifikace neustále
aktualizována.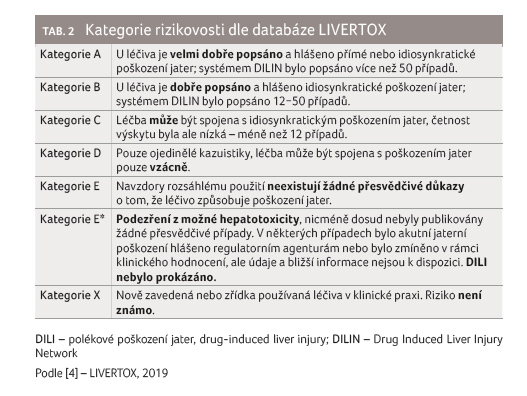
Pro zařazení léčiva je kromě četnosti výskytu rozhodující
především závažnost poškození. Ta se může značně lišit a zahrnovat podoby
od mírného, přechodného a asymptomatického zvýšení aktivity jaterních
enzymů v séru až po akutní selhání jater, které může ústit
v úmrtí či v potřebu transplantace jater. Hodnocení vychází
z pětibodové číselné stupnice.
- 1+, mírné poškození: zvýšené aktivity aminotransferáz v séru nebo alkalické fosfatázy nebo obojí, ale celková sérová koncentrace bilirubinu < 2,5 mg/dl a žádná koagulopatie (INR < 1,5);
- 2+, středně závažné poškození: zvýšené aktivity aminotransferáz nebo alkalické fosfatázy nebo obojí a celková sérová koncentrace bilirubinu > 2,5 mg/dl nebo koagulopatie (INR > 1,5) bez hyperbilirubinemie;
-
3+, střední až závažné poškození: zvýšené aktivity aminotransferáz nebo alkalických fosfatáz a celková sérová koncentrace bilirubinu > 2,5 mg/dl a hospitalizace v důsledku poškození jater;
- 4+, závažné poškození: zvýšené aktivity aminotransferáz nebo alkalických fosfatáz a sérová koncentrace bilirubinu > 2,5 mg/dl a alespoň jeden z následujících stavů:
- přetrvávající žloutenka,
- známky jaterní dekompenzace (INR > 1,5, ascites, encefalopatie),
- související poškození dalších orgánů;
Hepatotoxicita inhibitorů CDK4/6
Ze skupiny inhibitorů cyklin‑dependentních kináz 4
a 6 (CDK4/6) lze v současnosti využít palbociklib a ribociklib,
v dohledné době pak abemaciklib. Jedná se o léčiva, která významným
způsobem zlepšují prognózu pacientek s nádorem prsu (HER negativní).
Z databáze LIVERTOX vyplývá, že ribociklib se jeví na první pohled
stran hepatotoxicity rizikovější (skóre C) ve srovnání
s abemaciklibem a palbociklibem (oba skóre E*). Širší klinická
zkušenost s touto skupinou léčiv nicméně dosud chybí. V průběhu
předregistračních studií došlo při podávání ribociklibu ke zvýšení
aktivity ALT u 46 % subjektů
(vs. 36 %
v kontrolní skupině), k více než pětinásobnému zvýšení hodnot nad
horní limit normálního rozmezí (ULN) u 10 %
(vs. 1 %). I přes
elevaci hodnot jaterních enzymů byl průběh obvykle asymptomatický. Pouze
v 1 % případů se
rozvinulo jaterní poškození včetně projevů žloutenky. Ve všech případech
nicméně došlo k úpravě po ukončení léčby (3−5 měsíců). Hepatotoxicita
byla pozorována nejčastěji po 3−5 cyklech léčby ribociklibem. I když
jaterní histologie vykazovala znaky podobné autoimunitní hepatitidě,
imunoalergické a autoimunitní rysy nebyly přítomny. V případě
abemaciklibu bylo dosaženo více než pětinásobných hodnot ALT ve 3−5 %, k méně závažnému
zvýšení aktivity ALT došlo přesto rovněž asi u 40 % pacientů. Klinicky významné poškození jater
nicméně pozorováno nebylo. Podobně tomu bylo také při podávání palbociklibu.
Inhibitory CDK4/6 jsou značně metabolizovány v játrech (převážně cestou
CYP3A4), přesný mechanismus vzniku jaterního poškození není objasněn.
Diskutována je možnost přímého působení na hepatocyty, ale také vliv
případných toxických nebo imunogenních metabolitů [4].
Hepatotoxicita inhibitorů JAK
Buněčná signalizace
prostřednictvím Janusových kináz (JAK; popsány jsou 4 typy – JAK1/2/3
a TYK2) hraje jednu z klíčových rolí s účastí na správné
funkci imunitního systému nebo hematopoezy. V klinické praxi je dosud
využíván především jejich inhibiční vliv na zánětlivý proces a tvorbu
chemokinů v synoviálních fibroblastech (tofacitinib, baricitinib). Další
oblastí, na kterou v současnosti cílí, jsou idiopatické střevní
záněty (tofacitinib, filgotinib ve fázi klinického hodnocení).
Z dostupných inhibitorů JAK lze také využít ruxolitinib. Přestože je jeho
schválenou indikací myelofibróza či polycytemie, zajímavých výsledků bylo
dosaženo také v léčbě kožního onemocnění – chronické mukokutánní
kandidózy. Ta může být geneticky podmíněna mutací transkripčního faktoru STAT1,
který je v úzkém spojení právě s JAK. Výsledkem je porucha vývoje
subpopulace T lymfocytů (Th17) s klíčovou rolí v obraně proti
povrchovým kvasinkovým infekcím [25].
Indikační pole inhibitorů JAK se tedy stále rozšiřuje.
S ohledem na hepatotoxicitu je považována za nejvýznamnější
inhibice JAK2. I proto většina v současnosti zkoušených inhibitorů
JAK vykazuje selektivitu především vůči JAK1 a JAK3. Historicky je
největší zkušenost s ruxolitinibem. V rámci klinického zkoušení bylo
pozorováno zvýšení sérové aktivity ALT u 25 %
pacientů (ve větvi s placebem u 7 %),
hodnot nad pětinásobek ULN pak u 1,3 %.
Jako překvapující se tedy může zdát, že ruxolitinib spadá do kategorie C,
viz tabulku 3. Jedním z důvodů je, že
od uvedení na trh bylo publikováno několik zpráv o reaktivaci
hepatitidy B v průběhu 1−6 měsíců od zahájení terapie. Velmi
blízkého zvýšení hodnot aminotransferázy bylo dosaženo jak u tofacitinibu
(28−34 %, hodnoty nad
trojnásobkem ULN u 1−2 %),
tak u baricitinibu (17 %,
hodnoty nad pětinásobkem ULN u méně než 1 %).
Oběma látkám bylo přesto přiřknuto skóre E*. Důvodem může být skutečnost, že
od schválení tofacitinibu i baricitinibu a jejich uvedení
na trh nebyly publikovány zprávy o hepatotoxicitě nebo reaktivaci
hepatitidy B. V obou případech je považováno klinicky zjevné
poškození jater za velmi vzácnou komplikaci. Určitou roli může také sehrát
míra jaterní metabolizace, ta je dominantní u tofacitinibu
a ruxolitinibu. Naopak baricitinib je metabolizován játry pouze asi z 10 %, většina podané látky
není v těle metabolizována a je eliminována v nezměněné formě
močí [4].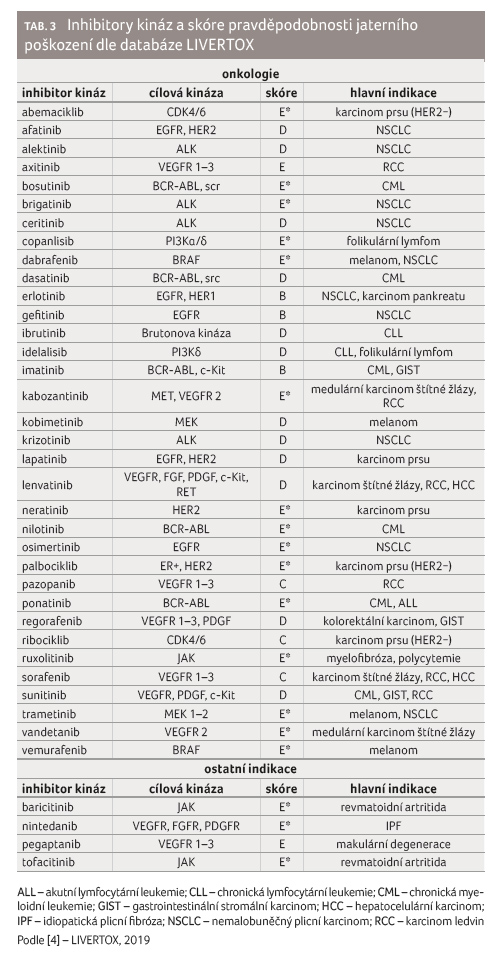
Hepatotoxicita inhibitorů ALK
Z dostupných
informací je zjevné, že hepatotoxicita je u inhibitorů anaplastické
lymfomové kinázy (anaplastic lymphoma kinase, ALK) typickým nežádoucím účinkem.
Léčiva této skupiny jsou používána v léčbě nemalobuněčného karcinomu plic
(non‑small cell lung carcinoma, NSCLC). Ačkoliv databáze LIVERTOX uvádí, že
při podávání krizotinibu se hepatotoxicita vyskytuje v porovnání
s ostatními inhibitory ALK (alektinib, brigatinib, ceritinib) častěji
a obvykle také s vážnějším průběhem, výsledky rozsáhlé metaanalýzy
z roku 2017 toto nepotvrzují [26]. Ta vycházela z dat 10
klinických studií (celkem 1 908 pacientů)
a prokázala četnější incidenci jaterní toxicity naopak u ceritinibu.
Poškození jater se nicméně v případě krizotinibu typicky objevuje během
několika dnů a týdnů od zahájení léčby. Zpravidla dochází
k prudké elevaci hodnot hepatocelulárních enzymů. Klinický obraz je
variabilní, od asymptomatického po závažný stav. Riziko významného
poškození jater a selhání jater je zvýšeno u pacientů s již
existující cirhózou nebo s jaterním poškozením. Především velké
předregistrační studie ukázaly zvýšení aktivity jaterních enzymů v séru u více
než poloviny pacientů léčených krizotinibem. Hodnoty pohybující se nad
pětinásobkem ULN pak u 6 %
pacientů. Zvýšení aktivity jaterních enzymů, obvykle bez žloutenky, či zvýšení
hodnot alkalické fosfatázy se obvykle objevilo po 4−12 týdnech léčby.
I přesto, že většina pacientů reagovala dobře na ukončení léčby, kdy
v průběhu 1−2 měsíců došlo k úpravě, vyskytly se také při léčbě
krizotinibem závažné případy s výraznou
elevací hodnot jaterních enzymů, s progresivní jaterní dysfunkcí,
koagulopatií, encefalopatií, a dokonce úmrtí. I to je důvodem, proč
se doporučuje pravidelné monitorování jaterních testů v intervalu
2−4 týdnů. Tak jako u většiny ki
příčina jaterního poškození není u krizotinibu dosud objasněna. Zbylé
inhibitory ALK vykazovaly rovněž časté zvýšení aktivity jaterních enzymů
(alektinib 50 %, ceritinib
20−50 %, brigatinib 40 %). Dle skóre rizikovosti patří alektinib,
ceritinib i krizotinib do shodné kategorie D, brigatinib
z důvodů dosud omezené zkušenosti do kategorie E* [4].
Hepatotoxicita inhibitorů EGFR
S předchozí kapitolou
má úzkou spojitost další skupina léčiv, která ovlivňuje buněčnou signalizaci
inhibicí receptoru pro epidermální růstový faktor (epidermal growth factor
receptor, EGFR). Tak jako je tomu u inhibitorů ALK, jsou i tato
léčiva využívána v terapii NSCLC. Předpokladem léčby je potvrzení
aktivační mutace EGFR. Do 1. generace patří reverzibilní inhibitory
gefitinib a erlotinib. Časem je s léčbou nicméně spojen vznik
rezistence na podkladě sekundární mutace (molekulární aberace). Za takové
situace se následně přistupuje ke 2. generaci inhibitorů EGFR, jimiž
jsou afatinib a nově dacomitinib. Tyto látky svou ireverzibilní
(kovalentní) vazbou úspěšnost léčby dále zvyšují. V případě průkazu mutace
T790M je používán osimertinib (3. generace). Hepatotoxicita je skupinovým
nežádoucím účinkem inhibitorů EGFR. Přesto se zdá, že poškození jater se
častěji a s vyšší intenzitou vyskytuje u gefitinibu (skóre B)
ve srovnání s erlotinibem (skóre B), afatinibem (skóre D)
i osimertinibem (skóre E*). U gefitinibu se dokonce jedná
o nejčastější nežádoucí účinek. V případě erlotinibu se jednalo
o kožní projevy, u afatinibu to byl průjem [27]. Případová studie
dokonce prokázala, že erlotinib byl účinný a současně lépe snášený jako
alternativa po ukončení léčby gefitinibem právě v důsledku
hepatotoxicity [28]. Ke zvýšení aktivity aminotransferáz v séru
dochází u 9−13 % pacientů
(dle některých studií až u 24 %), u 2−4 % pacientů dosahuje zvýšení hodnot pětinásobku ULN
[4,27]. K hepatocelulárnímu charakteru jaterního poškození
a ke zvýšení aktivity enzymů obvykle dochází v průběhu 1−2
měsíců léčby. K úpravě stavu rovněž za 1−2 měsíce po přerušení léčby.
Většina případů je spíše asymptomatických. Klinicky zjevné poškození jater se
žloutenkou je sice vzácné, přesto byly hlášeny i případy závažné
a fatální hepatotoxicity. Databáze LIVERTOX uvádí v případě
erlotinibu četnost výskytu zvýšení hodnot nad pětinásobek ULN u více než
10 % pacientů. Tato hodnota se ale zdá být
zkreslující, neboť vychází z údajů, kdy byl současně podán erlotinib
v kombinaci s konvenční chemoterapií. Při sledování hepatotoxického
potenciálu samotného erlotinibu byla pozorována výrazně nižší četnost elevace
hodnot AST i ALT (3,3 % a 2,2 %, stupeň ≥ 3) [27]. S ještě menší
četností se objevuje zvýšení aktivit aminotransferáz nad pětinásobek ULN
u afatinibu (1−2 % pacientů).
Vyskytly se i případy náhlého a klinicky závažného poškození jater,
což naznačuje, že podkladem může být imunitní reakce. Přechodné zvýšení hodnot
jaterních enzymů, které není v průběhu léčby neobvyklé, však ukazuje spíše
na přímou hepatotoxicitu [4].
Hepatotoxicita KI ovlivňující angiogenezi
U nádorových buněk dochází v důsledku jejich nadměrného
růstu a související potřeby kyslíku a živin ke zvýšené produkci
vaskulárního endoteliálního růstového faktoru (vascular endothelial growth
factor, VEGF). Ten vazbou na své receptory (VEGFR) zásadním způsobem
ovlivňuje tvorbu nových cév. Kvalita takto nově vzniklých cév není
plnohodnotná, což je mimo jiné jednou z příčin špatného průniku chemoterapie
k nádorovým buňkám. Tato indikačně heterogenní skupina KI potlačuje tvorbu
nových cév, současně dochází také ke změnám cév již vytvořených. To
především v případě nádorových buněk umožňuje lepší průnik léčiv, následně
zpomalení až zastavení růstu. Inhibice angiogeneze je rovněž využíváno při
léčbě idiopatické plicní fibrózy (nintedanib).
Z pohledu hepatotoxicity jsou významnými KI především
regorafenib, sorafenib a sunitinib. Dle databáze LIVERTOX spadají
do třídy B, což znamená, že u nich byly dobře popsány případy
jaterního poškození. Při podávání regorafenibu se vyskytuje zvýšení aktivity
sérových aminotransferáz až u 45 % pacientů,
zvýšení hodnot nad pětinásobek ULN je pozorováno u 3−6 % pacientů. Bylo dokonce
hlášeno několik případů závažného jaterního selhání, a to včetně úmrtí.
Poškození jater způsobené regorafenibem se může
manifestovat s výraznou variabilitou [4]:
- rozvoj akutní jaterní nekrózy (v průběhu několika dnů po zahájení léčby), stav je doprovázen vysokými hodnotami sérové aminotransferázy a laktátdehydrogenázy, prodloužením INR (mezinárodního normalizovaného poměru) a příznaky selhání jater;
- jaterní poškození pod obrazem akutní virové hepatitidy;
- jaterní poškození na podkladě autoimunitní a imunoalergické reakce (méně časté);
- sinusoidální obstrukční syndrom, pseudocirhóza s výraznou jaterní nodularitou a ascitem (vzácně);
- hyperamonemické kóma (v průběhu několika dnů až týdnů), jedná se o závažný stav vzniklý v důsledku léčby multikinázovými inhibitory − regorafenibem, sunitinibem, sorafenibem (ojedinělé epizody).
Při léčbě sorafenibem lze očekávat zvýšení aktivity
aminotransferáz přibližně u poloviny pacientů, hodnoty převyšující
pětinásobek ULN jsou nicméně popisovány pouze u 1−3 % pacientů. Přesto existuje řada případů
závažného poškození jater včetně úmrtí. Latence akutní hepatotoxicity
(hepatocelulární typ) se pohybovala v rozmezí od několika dnů
do zhruba osmi týdnů od zahájení léčby. Po ukončení terapie
obvykle nastala rychlá úprava stavu [4].
Z hlediska hepatotoxicity stojí za upozornění
možná interakce mezi KI a paracetamolem. Na tuto interakci je
pamatováno například v souhrnné informaci o přípravku imatinib,
v případě sorafenibu tomu tak však není. Sorafenib inhibuje aktivitu UDP‑glukuronosyltransferázy,
což může vést k mírné hyperbilirubinemii, ale také může zvyšovat toxicitu
současně podávaného paracetamolu (snížení schopnosti glukuronidace). Tato
potenciálně riziková interakce byla popsána také u dasatinibu
a imatinibu. Menší měrou je ovlivněna aktivita UDP‑glukuronosyltransferázy
také u axitinibu, erlotinibu, gefitinibu, lapatinibu, nilotinibu
a vandetanibu [29]. Tak jako v případě regorafenibu byly
i u sorafenibu hlášeny případy hyperamonemie
s následnou encefalopatií s latencí 1−3 týdny [4].
Registrační studie se sunitinibem také potvrdily dle
očekávání zvýšení aktivity aminotransferáz (39 %
pacientů), zvýšení hodnot nad pětinásobek ULN se vyskytlo pouze u 2−3 % pacientů. Poškození
jater v některých případech probíhalo pod obrazem akutní jaterní nekrózy.
Ačkoliv mechanismus vzniku hepatotoxicity není zcela objasněn, klinický obraz
vážného akutního poškození jater vzniklého v důsledku léčby sunitinibem
ukazuje na ischemické poškození [4].
Závěr
Závěrem je třeba
zdůraznit, že potenciál DILI u některých léčiv nebyl dosud plně rozpoznán.
Inhibitory kináz navíc představují novou skupinu léčiv. Přesto se cílená léčba
těmito přípravky stala již nedílnou součástí řady terapeutických postupů.
I proto se počet pacientů užívajících tuto skupinu léčiv neustále zvyšuje.
Většina informací stran hepatotoxicity vychází především z výsledků
předregistračních nebo případových studií. Detailní příčiny vzniku poškození
jater spíše chybějí nebo jsou znalosti o nich omezené. To je
v souladu s publikovanými závěry řady studií, které se soustředily
na hepatotoxicitu inhibitorů kináz. Nejčastěji zvažovaná mitochondriální
toxicita prokázala pouze omezenou schopnost predikce DILI. Další markery, které
by byly schopny predikovat jaterní poškození, jsou hledány.
Seznam použité literatury
- [1] Červený P. Polékové poškození jater. Prakt lékáren 2013; 9: 123–126.
- [2] Weng Z, Wang K, Li H, Shi Q. A comprehensive study of the association between drug hepatotoxicity and daily dose, liver metabolism, and lipophilicity using 975 oral medications. Oncotarget 2015; 6: 17031–17038.
- [3] Weng Z, Zhou P, Salminen WF. Green tea epigallocatechin gallate binds to and inhibits respiratory complexes in swelling but not normal rat hepatic mitochondria. Biochem Biophys Res Commun 2014; 443: 1097–1104.
- [4] National Institutes of Health – Databáze LIVERTOX [online], 2019. Dostupné na: https://livertox.nih.gov/
- [5] Robles Díaz M, Medina Caliz I, Stephens C. Biomarkers in DILI: One More Step Forward. Front Pharmacol 2016; 7: 267.
- [6] Shah RR, Morganroth J, Shah DR. Hepatotoxicity of Tyrosine Kinase Inhibitors: Clinical and Regulatory Perspectives. Drug Saf 2013; 36: 491−503.
- [7] Spragg CF, Budde LR, Briley LP. HLA DQA1_02:01 is a major risk factor for lapatinib induced hepatotoxicity in women with advanced breast cancer. J Clin Oncol 2011; 29: 667–673.
- [8] Sato Y, Fujimoto D, Shibata Y. Fulminant hepatitis following crizotinib administration for ALK positive nonsmall cell lung carcinoma. Jpn J Clin Oncol 2014; 44: 872–875.
- [9] Huang YS, An SJ, Chen ZH. Three cases of severe hepatic impairment caused by erlotinib. Br J Clin Pharmacol 2009; 68: 464–467.
- [10] Ridruejo E, Cacchione R, Villamil AG. Imatinib induced fatal acute liver failure. World J Gastroenterol 2007; 13: 6608–6611.
- [11] Klempner SJ, Choueiri TK, Yee E. Severe pazopanib induced hepatotoxicity: clinical and histologic course in two patients. J Clin Oncol 2012; 30: 264–268.
- [12] Price KE, Saleem N, Lee G. Potential of ponatinib to treat chronic myeloid leukemia and acute lymphoblastic leukemia. Onco Targets Ther 2013; 6: 1111–1118.
- [13] Gore ME, Szczylik C, Porta C. Safety and efficacy of sunitinib for metastatic renal cell carcinoma: an expanded access trial. Lancet Oncol 2009; 10: 757–763.
- [14] Mingard C, Paech F, Krähenbühl S. Mechanisms of toxicity associated with six tyrosine kinase inhibitors in human hepatocyte cell lines. J Appl Toxicol 2018; 38: 418−431.
- [15] Zhang J, Salminen A, Yang X. Effects of 31 FDA approved small molecule kinase inhibitors on isolated rat liver mitochondria. Arch Toxicol 2017; 91: 2921−2938.
- [16] Castellino S, O’Mara M, Koch K. Human metabolism of lapatinib, a dual kinase inhibitor: implications for hepatotoxicity. Drug Metab Dispos 2012; 40: 139–150.
- [17] Teo YL, Ho HK, Chan A. Formation of reactive metabolites and management of tyrosine kinase inhibitor induced hepatotoxicity: a literature review. Expert Opin Drug Metab Toxicol 2015; 11: 231–242.
- [18] Weng Z, Luo Y, Yang X. Regorafenib impairs mitochondrial functions, activates AMP activated protein kinase, induces autophagy, and causes rat hepatocyte necro-sis. Toxicology 2015; 327: 10–21.
- [19] Xue T, Luo P, Zhu H. Oxidative stress is involved in Dasatinib induced apoptosis in rat primary hepatocytes. Toxicol Appl Pharmacol 2012; 261: 280–291.
- [20] Feng B, Xu JJ, Bi YA. Role of hepatic transporters in the disposition and hepatotoxicity of a HER2 tyrosine kinase inhibitor CP 724,714. Toxicol Sci 2009; 108: 492–500.
- [21] Sugiyama E, Umemura S, Nomura S. Impact of single nucleotide polymorphisms on severe hepatotoxicity induced by EGFR tyrosine kinase inhibitors in patients with non small cell lung cancer harboring EGFR mutations. Lung Cancer 2015; 90: 307–313.
- [22] Takimoto T, Kijima T, Otani Y. Polymorphisms of CYP2D6 gene and gefitinib induced hepatotoxicity. Clin Lung Cancer 2013; 14: 502–507.
- [23] Paech F, Bouitbir J, Krähenbühl S. Hepatocellular Toxicity Associated with Tyrosine Kinase Inhibitors: Mitochondrial Damage and Inhibition of Glycolysis. Front Pharmacol 2017; 8: 367.
- [24] Lai GM, Yan SL, Chang CS. Hepatitis B reactivation in chronic myeloid leukemia patients receiving tyrosine kinase inhibitor. World J Gastroenterol 2013; 19: 1318–1321.
- [25] Bloomfield M, Kanderová V, Pračková Z. Utility of Ruxolitinib in a Child with Chronic Mucocutaneous Candidiasis Caused by a Novel STAT1 Gain of Function Mutation. J Clin Immunol 2018; 38: 589−601.
- [26] Liu B, Yuan M, Sun Y. Incidence and risk of hepatic toxicities associated with anaplastic lymphoma kinase inhibitors in the treatment of non small cell lung cancer: a systematic review and meta analysis. Oncotarget 2017; 9: 9480–9488.
- [27] Takeda M, Nakagawa K. Toxicity profile of epidermal growth factor receptor tyrosine kinase inhibitors in patients with epidermal growth factor receptor gene muta-tion positive lung cancer. Mol Clin Oncol 2016; 6: 3–6.
- [28] Takeda M, Okamoto I, Tsurutani J. Clinical impact of switching to a second EGFR TKI after a severe AE related to a first EGFR TKI in EGFR mutated NSCLC. Jpn J Clin On-col 2012; 42: 528–533.
- [29] Liu Y, Ramírez J, Ratain MJ. Inhibition of paracetamol glucuronidation by tyrosine kinase inhibitors. Br J Clin Pharmacol 2011; 71: 917–920.
- [30] Bunchorntavakul C, Reddy R. Drug Hepatotoxicity: Newer Agents. Clin Liver Dis 2017; 21: 115−134.