Inzulinová rezistence a možnosti jejího ovlivnění
Inzulinová rezistence znamená poruchu v účinku inzulinu a definujeme ji jako stav, při němž normální plazmatické hladiny inzulinu vyvolávají nižší biologickou odpověď v organismu. Klinicky je hlavním projevem porucha v oblasti metabolismu glukózy, i když rezistence se projevuje i v dalších oblastech účinků inzulinu, jako je metabolismus tuků a bílkovin, proliferační a mitogenní efekt či vliv na sekreci vazoaktivních působků. Inzulinová rezistence je asociována s celou řadou patologických stavů, nejčastěji ji však potkáváme jako podklad metabolického syndromu, který výrazně zhoršuje prognózu nemocných a vede ke zvýšenému riziku akcelerace aterosklerózy, rozvoje kardiovaskulárních komplikací, zvýšení celkové mortality, morbidity a rizika vzniku některých nádorů. Inzulinovou rezistenci můžeme přímo ovlivňovat režimovými opatřeními (dieta, fyzická aktivita), ale také léky zlepšujícími inzulinovou senzitivitu (metformin, thiazolidindiony). Součástí léčby metabolického syndromu je však i ovlivnění jeho dalších klinických komponent, např. hypertenze, dyslipidémie nebo prokoagulačního stavu.
Úvod
Inzulinová rezistence znamená poruchu v účinku inzulinu a definujeme ji jako stav, při němž normální plazmatické hladiny inzulinu vyvolávají nižší biologickou odpověď v organismu. Klinicky je hlavním projevem porucha v oblasti metabolismu glukózy, i když rezistence se projevuje i v dalších oblastech účinků inzulinu, jako je metabolismus tuků a bílkovin, proliferační a mitogenní efekt či vliv na sekreci vazoaktivních působků.
Sekrece inzulinu a jeho účinky
Inzulin uvolňovaný z β-buněk Langerhansových ostrůvků pankreatu pulzní sekrecí působí v příčně pruhovaném svalu, tukové tkáni a játrech prostřednictvím specifických receptorů. Po vazbě inzulinu na inzulinový receptor dojde k autofosforylaci tyrozinkinázy, která je signálem pro kaskádu dalších reakcí. Ústřední molekulou, na niž se přenáší signál z aktivovaného receptoru, je substrát inzulinového receptoru (IRS). Jeho aktivace vede ke stimulaci fosfatidylinositol-3-kinázy (PI3K), která zprostředkovává metabolické účinky inzulinu – vede ke zvýšení lipogeneze a proteosyntézy; stimulací glykogensyntázy vede ke zvýšenému ukládání glykogenu v játrech. Stimulací PI3K dochází rovněž k přesunu glukózových transportérů GLUT 4 z nitra buňky k jejímu povrchu, což je následováno otevřením transportních kanálů pro glukózu, a tak je umožněn vstup glukózy do buněk. Tímto mechanismem je regulována postprandiální glykémie v tkáních závislých na účinku inzulinu.
Uvedené děje jsou udržovány v rovnováze zpětnovazebnými okruhy, na nichž se podílejí vztahy mezi glykémií a inzulinem, inzulinem a hormony k inzulinu antagonistickými (glukagon, katecholaminy, růstový hormon, kortisol), ale také hormonální regulace vycházející z trávicí trubice (glukózo-dependentní inzulinotropní peptid GIP a somatostatin), která se uplatňuje zejména v postprandiální fázi.
Po několika hodinách lačnění, kdy chybí stimulace potravou, se sekrece inzulinu ustaluje na nízkých hodnotách (bazální sekrece). Probíhá v pulzním režimu, který udržuje senzitivitu cílových tkání k inzulinu, takže i při nízkých koncentracích inzulinu v plazmě (kolem 5–6 mU/l) je zachována dobrá vnímavost a odpovídavost cílových buněk. Po stimulaci jídlem se inzulin uvolňuje z β-buněk ze sekrečních granulí (časná fáze sekrece), dostává se k cílovým tkáním, které jsou citlivé. Trvá-li stimulus déle, přechází sekrece inzulinu do pozdní fáze. Déle trvající přísun většího množství inzulinu k cílové buňce vyvolává down regulaci inzulinových receptorů, která chrání buňku před přílišným poklesem glykémie. Děj probíhá velmi pružně a změny lze prokázat již po několika minutách. Při přechodu zpět do lačného stavu či až k hladovění se objevují pochody opačné, dochází k recyklaci a up regulaci inzulinových receptorů. U jedinců se zachovanou dobrou citlivostí k inzulinu jsou tyto děje velmi jemně regulovány [1].
Pokud dobře fungují výše uvedené vztahy a regulační mechanismy, je zachováno normální působení inzulinu. V případě narušení těchto vztahů dochází ke snížení účinku inzulinu a ke vzniku inzulinové rezistence. Jsou-li zachované regulační mechanismy, může být nastolena nová rovnováha, při níž zvýšené požadavky na inzulin podpoří jeho vyšší sekreci z β-buněk (= vzniká hyperinzulinémie), vyšší plazmatická hladina inzulinu potom zajistí jeho přijatelný účinek.
Inzulinová rezistence a metabolismus glukózy
Kompenzovaný vztah inzulinová rezistence–kompenzatorní hyperinzulinémie zachovává normální transport glukózy do svalové i tukové buňky a tím i normální glukózovou toleranci. K této situaci běžně dochází při přejídání, zejména za současného omezení fyzické aktivity, které vede k přibývání tělesné hmotnosti. Jde o reverzibilní děj; úprava životosprávy, pokles energetického příjmu a zvýšení fyzické aktivity může zcela upravit původní inzulinovou senzitivitu.
Inzulinová rezistence se manifestuje jednak v tukové a svalové tkáni, jednak v játrech. Relativní nedostatek inzulinu v rámci inzulinové rezistence se projeví neschopností potlačit glukoneogenezi v játrech, dochází ke zvýšené produkci glukózy z jater, na jejím odsunu z cirkulace potom závisí stupeň poruchy tolerance glukózy. Při současně přítomné periferní inzulinové rezistenci ve svalech a tukové tkáni se objevuje hyperglykémie nalačno, výše glykémie bývá výrazem stupně inzulinové rezistence.
Při rozvoji inzulinové rezistence sehrávají důležitou úlohu i tuky. Fyziologicky působí volné mastné kyseliny synergicky s glukózou jako významný stimulátor inzulinové sekrece. Při hladovění s poklesem hladiny volných mastných kyselin klesá i stimulační efekt glukózy na β-buňky, sekrece inzulinu klesá. Citlivost β-buňky a její sekreční odpověď závisí na oscilaci hladin glukózy a volných mastných kyselin. Krátkodobé zvýšení jejich hladiny tedy zvyšuje i sekreci inzulinu. Dochází-li však k dlouhodobému zvýšení hladin volných mastných kyselin, je v důsledku sekrečního podnětu přítomna hyperinzulinémie a hladina inzulinu je vyšší, než by odpovídalo stimulačnímu efektu samotné glukózy. V této situaci je naopak tlumena sekrece inzulinu stimulovaná glukózou, takže postupem času vede chronicky zvýšená hladina volných mastných kyselin ke snížení sekrece inzulinu. Zvýšený přísun volných mastných kyselin do periferních tkání současně vede k poklesu utilizace glukózy (kompetice v rámci Randlova cyklu) [1].
Počáteční hyperinzulinémie se pojí s rozvojem inzulinové rezistence, která se prohlubuje při ukládání tuků (především triglyceridů) do svalové tkáně, jater a β-buňky. Zdrojem volných mastných kyselin může být zvýšená konzumace nebo zvýšené uvolňování z viscerální tukové tkáně. Primárně inzulinotropní působení volných mastných kyselin na β-buňku chrání organismus před jejich nadměrným uvolňováním z tukové tkáně. Přetrvávající přísun volných mastných kyselin spolu s infiltrací β-buněk triglyceridy vede k jejich postupnému selhávání a ke zvýšené apoptóze, s následným útlumem nejen sekrece, ale i syntézy inzulinu (= lipotoxicita). Výsledkem je snížení plazmatické hladiny inzulinu, které prohlubuje defekty způsobené inzulinovou rezistencí v periferních tkáních.
Z kombinace těchto vlivů pak vyplývá možnost rozvoje diabetes mellitus 2. typu. Úloha volných mastných kyselin a poruchy metabolismu lipidů se jeví v patogenezi inzulinové rezistence, ale i v patogenezi diabetes mellitus 2. typu jako zcela zásadní [2].
Inzulinová rezistence a tuková tkáň
Vztah mezi inzulinovou rezistencí a obezitou byl potvrzen v longitudinálních studiích, nicméně pokračují diskuse, zda tuková depozita, viscerální tuková tkáň nebo subkutánní tuková tkáň mají v tomto vztahu důležitější úlohu. Ve studii IRAS (Insulin Resistance Atherosclerosis family Study) byl na 1500 dospělých hispánské a afroamerické národnosti prokázán jasný nezávislý vztah jak mezi viscerální, tak subkutánní adipozitou a inzulinovou rezistencí. Viscerální adipozita byla přitom více potentním prediktorem inzulinové rezistence než subkutánní adipozita. Akumulace viscerálního tuku zhoršovala inzulinovou senzitivitu a zvyšovala jaterní glukoneogenezi [3, 4].
Zdá se však také, že depozita tuku v játrech a kosterním svalstvu vedou v těchto orgánech ke vzniku inzulinové rezistence, která způsobuje zvýšenou glykémii nalačno (při akcelerované hepatální produkci glukózy) a postprandiální hyperglykémii (při sníženém zpracovávání glukózy ve svalech).
Inzulinová rezistence v tukové tkáni vede ke zvýšenému uvolňování volných mastných kyselin z adipocytů, protože hormon-senzitivní lipáza není nedostatečným účinkem inzulinu inhibována. Zdá se, že zvýšená hladina volných mastných kyselin je centrální složkou syndromu inzulinové rezistence, především z pohledu „overflow“ hypotézy. Je-li zásobní kapacita adipocytů překročena, dochází k „přetečení“, a tudíž k akumulaci volných mastných kyselin ve formě toxických ceramidů a sfingolipidů ve svalech, játrech, pankreatu a tepnách, kde způsobují klinické známky inzulinové rezistence. Plazmatické volné mastné kyseliny rovněž vedou k aktivaci zánětlivých cytokinů, zvýšení sekrece tumor nekrotizujícího faktoru a a růstových faktorů, což přispívá ke vzniku prozánětlivého stavu a k akceleraci aterosklerózy [5, 6].
Inzulinová rezistence, diabetes mellitus 2. typu a obezita
Stejně jako diabetes mellitus 2. typu (DM2), i obezita je spojena s těžkou inzulinovou rezistencí a kompenzatorní hyperinzulinémií [7]. Zatímco u DM2 je inzulinová rezistence způsobena z větší části vrozeným metabolickým defektem, u obezity může být inzulinová rezistence následkem excesivního energetického příjmu nebo i vrozenou poruchou v termogenezi nebo intermediárním metabolismu [8]. Bazálně i po hyperglykemizujícím stimulu vykazují obézní hyperinzulinémii, která koreluje se stupněm inzulinové rezistence, ve snaze zachovat normální glukózovou toleranci. Řada faktorů spojených s obezitou (zvýšené hladiny volných mastných kyselin, snížené hladiny adiponektinu a zvýšené hladiny adipocytokinů) je zodpovědná za indukci inzulinové rezistence u obézních. Stejně jako je tomu při rozvoji DM2, může vést zhoršení metabolického stavu k selhání schopnosti endogenní hyperinzulinémie plně kompenzovat inzulinovou rezistenci a dojde k porušení glukózové tolerance [9].
Recentní studie poukazují na zánětlivý stav tukové tkáně při zvyšující se tělesné hmotnosti a vzniku obezity, který může podporovat vznik inzulinové rezistence dalším mechanismem. Zvýšený body-mass index, zvětšené množství viscerálního tuku a inzulinová rezistence jsou spojeny se zvýšenou hladinou cirkulujících markerů zánětu, jako je C-reaktivní protein, prozánětlivé cytokiny a solubilní adhezní molekuly. Na zvířecích modelech byla prokázána zvýšená infiltrace tukové tkáně aktivovanými makrofágy při krmení zvířat „západní“ dietou [10]. Úbytek hmotnosti byl následně spojen i s redukcí prozánětlivého stavu. Tato pozorování vedla k názoru, že zánětlivé mediátory produkované tukovou tkání jsou klíčovým hráčem v rozvoji inzulinové rezistence a endoteliální dysfunkce, vedoucí k manifestaci DM2 a aterosklerózy [11].
Inzulinová rezistence a metabolický syndrom
Inzulinová rezistence je asociována s celou řadou patologických stavů, ale určitý stupeň inzulinové rezistence či její vystupňování můžeme pozorovat i v některých „fyziologických“ situacích, jako je puberta, gravidita, stárnutí či psychický stres (tab. 1).
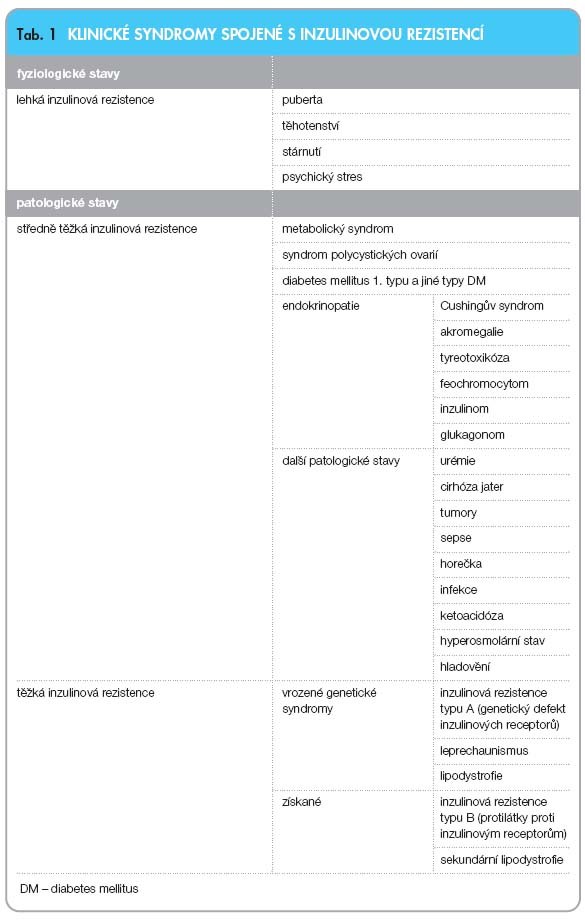 Kvantitativně nejvýznamnější část osob s projevy inzulinové rezistence tvoří osoby s metabolickým syndromem. Metabolický syndrom, též označovaný jako syndrom inzulinové rezistence, představuje soubor klinických, biochemických a humorálních odchylek, které vznikají v souvislosti s poruchou účinku inzulinu v metabolismu glukózy (obr. 1). Na sdružení obezity, diabetu a arteriální hypertenze poukazoval již Reaven a Kaplan jako na kombinaci, která výrazně zhoršuje prognózu nemocných a vede ke zvýšenému riziku akcelerace aterosklerózy, rozvoje kardiovaskulárních komplikací, zvýšení celkové mortality, morbidity a rizika vzniku některých nádorů (tumory tlustého střeva, prostaty, gynekologické nádory) [12]. Metabolický syndrom je onemocnění s polygenní dědičností, s expresí výrazně ovlivněnou zevními vlivy. V průběhu evoluce hrála významnou roli selekce jedinců s „úsporným metabolismem“, podporujícím ukládání i malého množství přijaté energie v období hladomoru do tukových zásob. Tento „thrifty genotyp“ se však stal extrémně nevýhodným při přechodu na životní styl s malým energetickým výdejem a velkým energetickým příjmem. Právě přejídání, obezita,
Kvantitativně nejvýznamnější část osob s projevy inzulinové rezistence tvoří osoby s metabolickým syndromem. Metabolický syndrom, též označovaný jako syndrom inzulinové rezistence, představuje soubor klinických, biochemických a humorálních odchylek, které vznikají v souvislosti s poruchou účinku inzulinu v metabolismu glukózy (obr. 1). Na sdružení obezity, diabetu a arteriální hypertenze poukazoval již Reaven a Kaplan jako na kombinaci, která výrazně zhoršuje prognózu nemocných a vede ke zvýšenému riziku akcelerace aterosklerózy, rozvoje kardiovaskulárních komplikací, zvýšení celkové mortality, morbidity a rizika vzniku některých nádorů (tumory tlustého střeva, prostaty, gynekologické nádory) [12]. Metabolický syndrom je onemocnění s polygenní dědičností, s expresí výrazně ovlivněnou zevními vlivy. V průběhu evoluce hrála významnou roli selekce jedinců s „úsporným metabolismem“, podporujícím ukládání i malého množství přijaté energie v období hladomoru do tukových zásob. Tento „thrifty genotyp“ se však stal extrémně nevýhodným při přechodu na životní styl s malým energetickým výdejem a velkým energetickým příjmem. Právě přejídání, obezita, 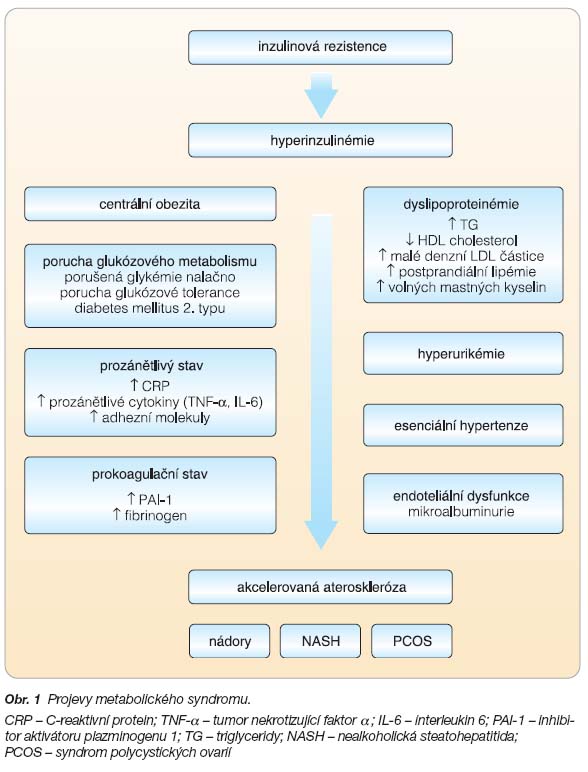 nedostatek fyzické aktivity, nevhodné složení stravy, stres, kouření a některé léky jsou významnými induktory inzulinové rezistence [13]. Prevalence metabolického syndromu se v naší populaci pohybuje kolem 25–30 %. Projevy metabolického syndromu jsou velmi pestré a jsou podmíněny širokou škálou účinků inzulinu. Inzulinová rezistence však nemusí být plně vyjádřena ve všech oblastech účinku inzulinu. Vedle inzulinové rezistence a ji doprovázející hyperinzulinémie je součástí metabolického syndromu centrální obezita, esenciální hypertenze, porucha metabolismu glukózy, dyslipoproteinémie, hyperurikémie, porucha hemokoagulace a fibrinolýzy, endoteliální dysfunkce a prozánětlivý stav. Postupem času se k syndromu inzulinové rezistence přiřazovaly pevněji či volněji další složky (nealkoholická steatohepatitida, hyperandrogenismus, hirsutismus, hyperhomocysteinémie a další) [14].
nedostatek fyzické aktivity, nevhodné složení stravy, stres, kouření a některé léky jsou významnými induktory inzulinové rezistence [13]. Prevalence metabolického syndromu se v naší populaci pohybuje kolem 25–30 %. Projevy metabolického syndromu jsou velmi pestré a jsou podmíněny širokou škálou účinků inzulinu. Inzulinová rezistence však nemusí být plně vyjádřena ve všech oblastech účinku inzulinu. Vedle inzulinové rezistence a ji doprovázející hyperinzulinémie je součástí metabolického syndromu centrální obezita, esenciální hypertenze, porucha metabolismu glukózy, dyslipoproteinémie, hyperurikémie, porucha hemokoagulace a fibrinolýzy, endoteliální dysfunkce a prozánětlivý stav. Postupem času se k syndromu inzulinové rezistence přiřazovaly pevněji či volněji další složky (nealkoholická steatohepatitida, hyperandrogenismus, hirsutismus, hyperhomocysteinémie a další) [14].
Klinické a biochemické projevy metabolického syndromu zvyšují riziko rozvoje aterosklerózy. Řada epidemiologických studií prokázala rizikovost jednotlivých složek metabolického syndromu a řada intervenčních studií naopak prokázala snížení celkové a kardiovaskulární mortality ovlivněním složek metabolického syndromu, především dyslipidémie (studie 4S, HPS, VA-HIT, CARE) a hypertenze (HOPE, HOT, Syst-Eur) [15].
Hyperinzulinémie a vaskulární efekty
Vedle zprostředkování metabolických účinků ve svalech, játrech a tukové tkáni plní inzulin funkci vaskulárního hormonu. Ve fyziologických dávkách u štíhlých osob způsobuje vazodilataci. Ale za podmínek inzulinové rezistence a hyperinzulinémie způsobuje inzulin paradoxně vazokonstrikci jako reakci na vazodilatační podněty (známka endoteliální dysfunkce). Byla rovněž prokázána zvýšená tvorba a snížená regrese aterosklerotických plátů, vyšší proliferace hladkých svalových buněk, zvýšená tvorba kolagenu, zvýšená aktivita LDL receptorů a zvýšení hladin růstových faktorů, což vše podporuje aterogenezi [16]. Tato data napovídají, že metabolická vazba mezi inzulinovou rezistencí/hyperinzulinémií a kardiovaskulárními rizikovými faktory může být vysvětlena hypotézou „tikajících hodin“ – hodiny pro aterosklerózu/kardiovaskulární komplikace začínají tikat při narození a jejich progrese závisí na vrozených dispozicích a hyperinzulinémii, které vystavují cévní systém inzulinové rezistenci a dalším kardiovaskulárním rizikovým faktorům ještě před vznikem hyperglykémie. Na druhou stranu, hodiny pro mikrovaskulární komplikace diabetu začínají tikat až při vzniku hyperglykémie [17].
Léčba osob s metabolickým syndromem
Osoby s metabolickým syndromem je možné léčit dvěma způsoby:
- přímým ovlivněním inzulinové rezistence;
- ovlivněním klinických projevů inzulinové rezistence s cílem snížit kardiovaskulární mortalitu a morbiditu
Přímé ovlivnění inzulinové rezistence
Léčbu vždy zahajujeme pokusem o ovlivnění inzulinové rezistence a jejích projevů změnou životního stylu, která zahrnuje dietní opatření a zvýšení fyzické aktivity. U nemocných s nadváhou a obezitou je základním léčebným prostředkem omezení energetického příjmu. Redukční dieta musí být spojena se změnou stravovacích návyků a kompletní změnou životního stylu. Jako podporu snížení energetického příjmu lze použít antiobezitika – sibutramin, orlistat. Nezbytnou součástí změny životního stylu je i zanechání kouření, které přímo zvyšuje inzulinovou rezistenci a zároveň je nezávislým rizikovým faktorem kardiovaskulárních onemocnění. Inzulinová rezistence klesá také v souvislosti se zlepšením kompenzace diabetu, nezávisle na způsobu jeho léčby. Všechna výše uvedená data však poukazují na to, že jako přídavek k dietě, cvičení, redukci hmotnosti a agresivní léčbě jednotlivých složek metabolického syndromu bychom měli začít co nejdříve ovlivňovat základní metabolický defekt, inzulinovou rezistenci, pomocí léků zvyšujících inzulinovou senzitivitu (metformin a thiazolidindiony).
Metformin
Metformin je již po čtyři desetiletí s úspěchem používán v léčbě nemocných s diabetem mellitem 2. typu. Metformin aktivuje enzym adenozinmonofosfát-proteinkinázu, který je klíčovým buněčným regulátorem glukózového a lipidového metabolismu a je odpovědný za citlivost buněk k působení inzulinu tím, že ovlivňuje inzulinovou signální cestu [18]. Metformin rovněž zvyšuje aktivitu glukózových transportérů GLUT 4, translokaci GLUT 1, normalizuje aktivitu enzymatického systému v intracelulární inzulinové signální kaskádě a stimuluje tyrozinkinázovou aktivitu intracelulárních b-podjednotek inzulinových receptorů. Výsledkem je antihyperglykemické působení na podkladě snížení jaterní produkce glukózy a zvýšení zpracovávání glukózy ve svalech.
O škodlivém vlivu hyperglykémie (glukotoxicita) a zvýšených hladin volných mastných kyselin (lipotoxicita) na sekreční činnost β-buněk pankreatických ostrůvků existuje dostatek důkazů. Prolongovaná expozice zvýšeným hladinám volných mastných kyselin vede k redukci glukózou stimulované sekrece inzulinu, k supresi biosyntézy proinzulinu a ke ztrátě β-buněk apoptózou. Ostrůvkové buňky diabetiků 2. typu vykazují tedy vážné funkční změny a poruchu v přežívání. Obě tyto situace mohou být zlepšeny, až téměř normalizovány metforminem, a to jak zprostředkovaně, redukcí oxidačního stresu, tak především přímým protektivním vlivem metforminu na lidské β-buňky [19].
Inzulinová rezistence je spojena s endoteliální dysfunkcí a poruchou sekrece vazoregulačních a koagulaci regulujících substancí, které endotelové buňky produkují. Jednou z těchto substancí je inhibitor plazminogenového aktivátoru 1 (PAI-1), hlavní antifibrinolytická komponenta v cirkulaci, která stoupá při inzulinové rezistenci. Metformin (v závislosti na dávce) snižuje zvýšené hodnoty PAI-1 o 43–100 %, čímž výrazně snižuje riziko trombogenních komplikací u inzulinorezistentních jedinců [17]. Metformin má přímý ochranný vliv proti poškození endotelových buněk hyperglykémií, čímž může výrazně ovlivnit výskyt makrovaskulárních komplikací u pacientů s DM2. Jako velice významná je v tomto ohledu hodnocena práce Detailleho a kol. [20]. Autoři sledovali možnost preventivního působení metforminu na apoptózu endotelových buněk vystavených vysokým koncentracím glukózy. Prokázali nejenom ochranný účinek metforminu, ale odhalili i mechanismus, jakým metformin chrání endotelové buňky před poškozením oxidačním stresem vyvolaným hyperglykémií. Metformin přímo ovlivňuje mitochondriální PTP (permeability transition pore), kanálky citlivé na oxidační stres, jejichž otevření znamená buněčnou smrt. Metformin rovněž snižuje tvorbu volných kyslíkových radikálů.
Thiazolidindiony
Inzulinové senzitizéry (glitazony, thiazolidindiony) jsou skupinou perorálních antidiabetik, účinných látek určených k léčbě DM2. V současné době jsou na trhu rosiglitazon a pioglitazon. Oba preparáty zlepšují inzulinovou senzitivitu. Jsou vysoce selektivními a potentními agonisty PPAR-g (peroxisome proliferator-activated receptor g), jaderných receptorů zapojených v lipidovém a sacharidovém metabolismu. PPAR-g se nacházejí v tkáních klíčových pro účinek inzulinu, jsou exprimovány v tukové tkáni, tlustém střevě, hematopoetických buňkách, v ledvinách, játrech, tenkém střevě a v kosterním svalu. Po vazbě thiazolidindionu na PPAR se vytvoří makromolekulární komplex schopný vázat se na určité sekvence DNA (tzv. thiazolidin-responzivní geny). 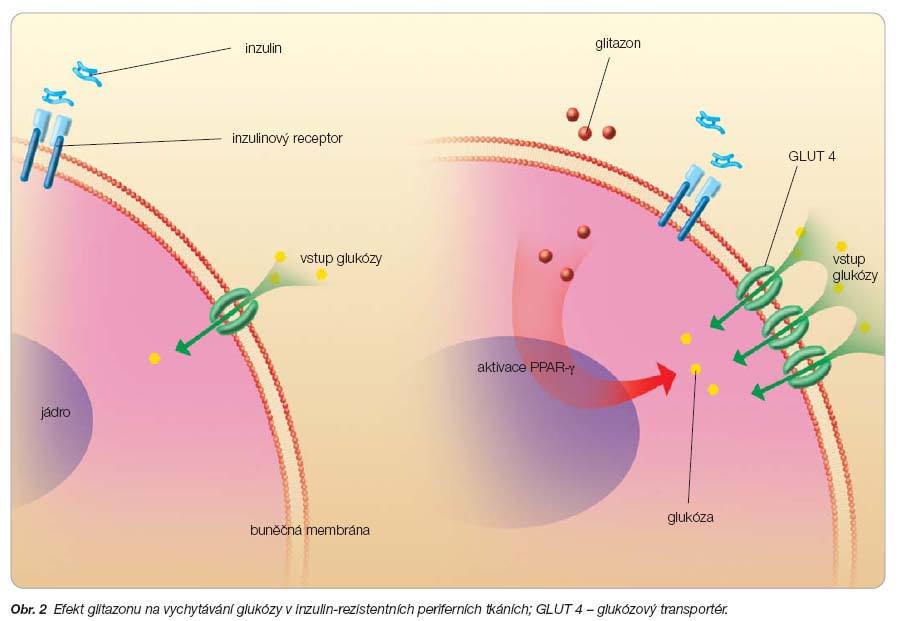 Celý děj ústí ve zvýšení transkripce specifických genů. Thiazolidindiony způsobují diferenciaci tukové tkáně do malých adipocytů, které jsou vůči inzulinu senzitivnější než velké adipocyty, a proto uvolňují do oběhu méně volných mastných kyselin, více adiponektinu a méně TNF-a, rezistinu a leptinu (tj. substancí, které mají vliv na inzulinovou rezistenci a také na dysfunkci endotelu). Receptor PPAR-g moduluje rovněž expresi genů, které produkují klíčové proteiny zapojené v regulaci produkce glukózy játry a vychytávání a skladování glukózy (přenašeč GLUT 4) v kosterních svalech a tukové tkáni (obr. 2).
Celý děj ústí ve zvýšení transkripce specifických genů. Thiazolidindiony způsobují diferenciaci tukové tkáně do malých adipocytů, které jsou vůči inzulinu senzitivnější než velké adipocyty, a proto uvolňují do oběhu méně volných mastných kyselin, více adiponektinu a méně TNF-a, rezistinu a leptinu (tj. substancí, které mají vliv na inzulinovou rezistenci a také na dysfunkci endotelu). Receptor PPAR-g moduluje rovněž expresi genů, které produkují klíčové proteiny zapojené v regulaci produkce glukózy játry a vychytávání a skladování glukózy (přenašeč GLUT 4) v kosterních svalech a tukové tkáni (obr. 2).
Vedle vlivu na snížení inzulinové rezistence ale vykazují thiazolidindiony řadu dalších účinků [21]. Třebaže thiazolidindiony nemodulují přímo sekreci inzulinu β-buňkami, zlepšením inzulinové rezistence (a rovněž stimulací receptorů PPAR-g přítomných v pankreatu) mohou zajistit ochranu inzulinové sekrece a pravděpodobně i „zpoždění“ v nástupu poklesu funkce β-buněk. 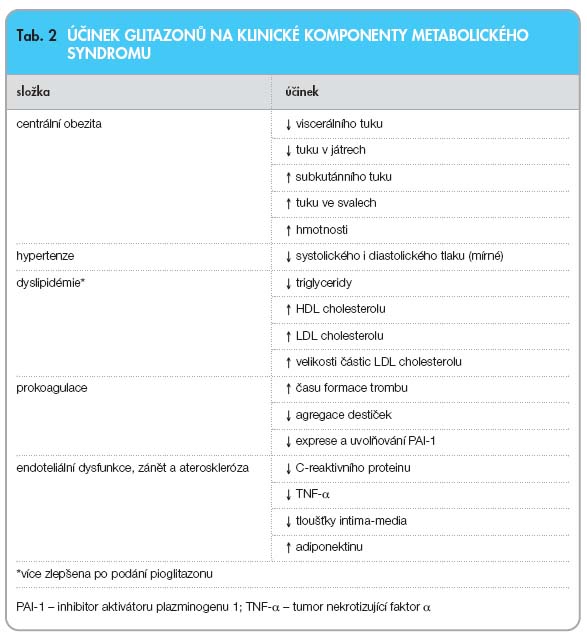 Thiazolidindiony svým účinkem ovlivňují celou řadu komponent metabolického syndromu (tab. 2). Vykazují příznivé efekty na plazmatické lipidové spektrum. Snižují hladinu volných mastných kyselin a signifikantně zvyšují hladiny HDL cholesterolu. Jejich účinek na hladinu triglyceridů a LDL cholesterolu je rozdílný, pravděpodobně způsobený větší selektivitou rosiglitazonu k PPAR-g. Zatímco při léčbě pioglitazonem dochází k poklesu hladiny triglyceridů, rosiglitazon hladinu triglyceridů zvyšuje. Obě léčiva sice zvyšují koncentraci LDL cholesterolu, avšak snižují podíl malých denzních částic LDL cholesterolu. Důležitým jevem a pravděpodobně i jedním z podkladů metabolického syndromu a inzulinové rezistence je akumulace tuku ve viscerální oblasti. Thiazolidindiony způsobují posun v distribuci tuku směrem z viscerálních oblastí do podkožních tukových zásob. Ačkoliv se celkový obsah tuku v těle mírně zvyšuje, dochází ke snížení inzulinové senzitivity v důsledku menšího množství metabolicky aktivních produktů viscerální tukové tkáně. Současně
Thiazolidindiony svým účinkem ovlivňují celou řadu komponent metabolického syndromu (tab. 2). Vykazují příznivé efekty na plazmatické lipidové spektrum. Snižují hladinu volných mastných kyselin a signifikantně zvyšují hladiny HDL cholesterolu. Jejich účinek na hladinu triglyceridů a LDL cholesterolu je rozdílný, pravděpodobně způsobený větší selektivitou rosiglitazonu k PPAR-g. Zatímco při léčbě pioglitazonem dochází k poklesu hladiny triglyceridů, rosiglitazon hladinu triglyceridů zvyšuje. Obě léčiva sice zvyšují koncentraci LDL cholesterolu, avšak snižují podíl malých denzních částic LDL cholesterolu. Důležitým jevem a pravděpodobně i jedním z podkladů metabolického syndromu a inzulinové rezistence je akumulace tuku ve viscerální oblasti. Thiazolidindiony způsobují posun v distribuci tuku směrem z viscerálních oblastí do podkožních tukových zásob. Ačkoliv se celkový obsah tuku v těle mírně zvyšuje, dochází ke snížení inzulinové senzitivity v důsledku menšího množství metabolicky aktivních produktů viscerální tukové tkáně. Současně 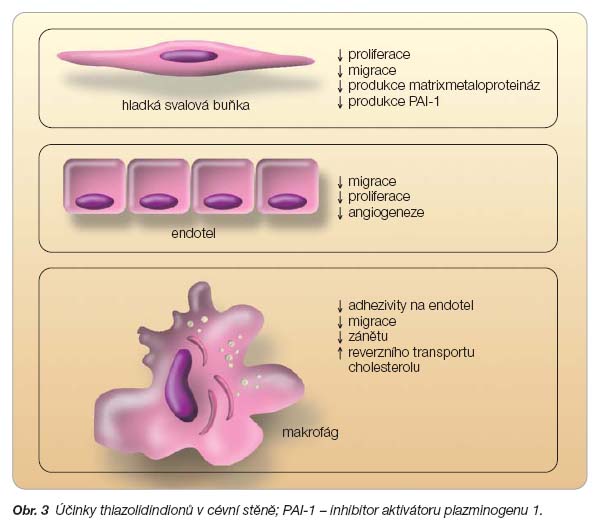 snížená hladina volných mastných kyselin vede ke vzniku zdravější tukové buňky, která lépe odpovídá na inzulin zvýšením vychytávání glukózy a supresí lipolýzy [22, 23]. Thiazolidindiony vykazují významný protizánětlivý a antiaterogenní efekt v cévní stěně (obr. 3), v průběhu léčby dochází ke snížení sérových hladin C-reaktivního proteinu a matrixmetaloproteináz, což vede ke zlepšení stability plátu. Thiazolidindiony modulují rovněž zánětlivý proces v cévní stěně, a ovlivňují tím skladbu aterosklerotického plátu. Léčba thiazolidindiony vede ke snížení tloušťky intima-media (IMT), markeru preklinické aterosklerózy, měřeného pomocí ultrazvuku s vysokou rozlišovací schopností. Ovlivněním proliferace hladkých svalových buněk cévní stěny brání léčba thiazolidindiony rovněž vzniku restenóz po koronárních intervencích [24, 25].
snížená hladina volných mastných kyselin vede ke vzniku zdravější tukové buňky, která lépe odpovídá na inzulin zvýšením vychytávání glukózy a supresí lipolýzy [22, 23]. Thiazolidindiony vykazují významný protizánětlivý a antiaterogenní efekt v cévní stěně (obr. 3), v průběhu léčby dochází ke snížení sérových hladin C-reaktivního proteinu a matrixmetaloproteináz, což vede ke zlepšení stability plátu. Thiazolidindiony modulují rovněž zánětlivý proces v cévní stěně, a ovlivňují tím skladbu aterosklerotického plátu. Léčba thiazolidindiony vede ke snížení tloušťky intima-media (IMT), markeru preklinické aterosklerózy, měřeného pomocí ultrazvuku s vysokou rozlišovací schopností. Ovlivněním proliferace hladkých svalových buněk cévní stěny brání léčba thiazolidindiony rovněž vzniku restenóz po koronárních intervencích [24, 25].
U 6–9 % pacientů léčených thiazolidindiony se objevují mírné až středně závažné otoky především dolních končetin. Thiazolidindiony vyvolávají retenci tekutin, která může vést ke vzniku nebo k prohloubení již přítomného srdečního selhání. Mechanismus vzniku otoku je nejasný, ale pravděpodobně souvisí s retencí tekutin a se zvýšením cévní permeability. U pacientů by měly být sledovány možné symptomy srdečního selhávání, přírůstek hmotnosti nebo edémy, zvláště pokud mají sníženou srdeční rezervu.
Velmi častým a pravděpodobně limitujícím nežádoucím účinkem léčby thiazolidindiony je zvyšování tělesné hmotnosti. Průměrný přírůstek hmotnosti tvoří asi 2–3 kg, ale u některých jedinců může být i významně větší. Objeví se především v prvním roce léčby. Příčina tkví zřejmě v souhrnu několika mechanismů: podpora tvorby tukové tkáně a současně i redistribuce tuku z viscerální oblasti do oblasti podkoží, retence tekutin a pravděpodobně i účinek zlepšení metabolické kompenzace.
V roce 2007 došlo k výraznému rozbouření hladiny zájmu o léčbu thiazolidindiony, především v souvislosti se zveřejněním metaanalýzy studií s rosiglitazonem. Výsledky ukazují, že pacienti léčení rosiglitazonem mají lehce vyšší riziko vzniku infarktu myokardu a úmrtí z kardiovaskulárních příčin. Prakticky okamžitě se rozvinula vášnivá diskuse, zda má rosiglitazon dostačující poměr risk/benefit a jak přistupovat k jeho preskripci. Jedná se o preparát ovlivňující genovou expresi, s poměrně komplexním účinkem na různých úrovních. Vývoj celé řady látek ze skupiny agonistů PPAR-g byl zastaven pro toxicitu. Jenomže vzhledem k rozdílnosti jednotlivých látek stran afinity k receptoru a ovlivnění rozdílných genů nelze hovořit o tzv. class efektu (skupinovém účinku). Pioglitazon se ukázal být velmi účinným i ve snížení kardiovaskulárních cílů ve studii PROactive, a ani recentně zveřejněná interim analýza probíhající studie Rosiglitazone Evaluated for Cardiac Outcomes and Regulation of Glycemia in Diabetes (RECORD, designovaná speciálně pro sledování kardiovaskulárního rizika) s rosiglitazonem nevychází negativně (bude dokončena v roce 2009). Prozatím je doporučeno nepodávat thiazolidindiony pacientům, pokud mají v anamnéze koronární onemocnění nebo jsou ohroženi vysokým rizikem vzniku tohoto onemocnění. Určitě má však význam důkladně ošetřit rizikové faktory kardiovaskulárních onemocnění (především je vhodná léčba statiny a antiagregancii).
Závěr
Inzulinová rezistence, především v klinickém spojení s metabolickým syndromem, je závažný stav, který jednoznačně zhoršuje prognózu nemocných a zvyšuje riziko akcelerace aterosklerózy, rozvoje kardiovaskulárních komplikací, celkové mortality, morbidity a riziko vzniku některých nádorů. Jako přídavek k dietě, cvičení, redukci hmotnosti a agresivní léčbě jednotlivých složek metabolického syndromu bychom měli začít co nejdříve ovlivňovat základní metabolický defekt – inzulinovou rezistenci – pomocí léků zvyšujících inzulinovou senzitivitu; cílem jejich podávání je co nejvíce zlepšit prognózu nemocných.
Seznam použité literatury
- [1] Škrha J. Patogeneze inzulinové rezistence. Vnitřní lékařství 2003; 49: 894–899.
- [2] Shafrir E, Raz I. Diabetes: mellitus or lipidus? Diabetologia 2003; 46: 433–440.
- [3] Castaldelli A, Miyazaki Y, Pettiti M, et al. Metabolic effects of visceral fat accumulation in type 2 diabetes. J Clin Endocrin Metab 2002; 87: 5098–5103.
- [4] Wagenknecht LE, Langefeld CD, Scherzinger AL, et al. Insulin sensitivity, insulin secretion and abdominal fat: the Insulin Resistance Atherosclerosis (IRAS) family study. Diabetes 2003; 52: 2490–2496.
- [5] Castaldelli A, Ferrannini E, Miyazaki Y, et al. Betacell dysfunction and glucose intolerance: results from San Antonio metabolism (SAM) study. Diabetologia 2004; 47: 31–39.
- [6] Kashyap S, Belfort R, Castaldelli A, et al. Chronically elevated free fatty acids concentrations produce a decrease in insulin secretion with no change in insulin sensitivity in individuals with heavy family history of T2DM. Diabetes 2003; 52: 2461–2474.
- [7] DeFronzo RA. Lilly lecture. The triumvirate: betacell, muscle, liver. A collusion responsible for NIDDM. Diabetes 1988; 37: 667–687.
- [8] DeFronzo RA, Bonadonna RC, Ferrannini E. Pathogenesis of NIDDM. A balanced overview. Diabetes Care 1992; 15: 318–368.
- [9] Bonadonna RC, Groop L, Kraemer N. Obesity and insulin resistance in humans: a dose-response study. Metabolism 1990; 39: 452–459.
- [10] Xu H, Barnes GT, Yang Q, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 2003; 112: 1821–1830.
- [11] Esposito K, Pontillo A, Di Palo C, et al. Effect of weight loss and lifestyle changes on vascular inflammatory marker in obese women: a randomized trial. JAMA 2003; 289: 1799–1804.
- [12] Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 1988; 37: 1595–1607.
- [13] Bloomgarden ZT. American Association of Clinical Endocrinologists (AACE). Consensus Conference on the insulin resistance syndrome. Diabetes Care 2003; 26: 933–939.
- [14] Svačina Š, Owen K, Bretschneiderová A. Syndrom inzulinové rezistence. Praha, Triton 2003.
- [15] Pelikánová T. Inzulinová rezistence a metabolický syndrom. Interní medicína pro praxi 2003; 10: 491–495.
- [16] Steinberg HO, Baron AD. Vascular function, insulin resistance and fatty acids. Diabetologia 2002; 45: 623–634.
- [17] Haffner SM, Stern MP, Hazuda HP, et al. Cardio-vascular risk factors in confirmed prediabetic individuals. Does the clock for coronary heart disease start ticking before the onset of clinical diabetes? JAMA 1990; 263: 2893–2898.
- [18] Garber AJ. Metformin and other biguanides: Pharmacology and therapeutic usage. In Int Textbook of Diabetes Mellitus, Third Edition, edited by DeFronzo RA, Ferrannini E, et al. John Wiley&Sons Ltd., 2004, 851–869.
- [19] Leclerc I, et al. Metformin, but not leptin, regulates AMP-activated protein kinase in pancreatic islets: impact on glucose-stimulated insulin secretion. Am J Physiol Endocrinol Metab 2004; 286: E1023–E1031.
- [20] Detaille D, Guigas B, Chauvin C, et al. Metformin prevents high-glucose-induced endothelial cell death through a mitochondrial permeability transition-dependent process. Diabetes 2005; 54: 2179–2187.
- [21] Bailey CJ. New drugs for the treatment of diabetes mellitus. In: Alberti MG (Ed.). International textbook of diabetes mellitus. John Wiley and Sons; 1997: 865–881.
- [22] Bays H, Mandarino L, DeFronzo R. Role of the adipocyte, free fatty acids, and ectopic fat in pathogenesis of type 2 diabetes mellitus: peroxisomal proliferator – activated receptor agonists provide a rational therapeutic approach. J Clin Endocrinol Metab 2004; 89: 463–478.
- [23] Komers R, Vrána A. Thiazolidinediones – tools for the research of metabolic syndrom X. Physiol Res 1998; 47: 215–225.
- [24] Elte JWF, Bliklé JF. Thiazolidindiones for the treatment of type 2 diabetes. European Journal of Internal Medicine 2007; 18: 18–25.
- [25] Langenfeld MR, Forst T, Hohnberg C, et al. Pioglitazone decreases carotid intima-media thickness independenty of glycemic control in patients with type 2 diabetes mellitus. Results from a controlled randomized study. Circulation 2005; 111: 2525–2531.