Léčba idiopatické plicní fibrózy
Idiopatická plicní fibróza (IPF) je vzhledem ke své obtížné léčitelnosti a prognóze nejzávažnějším reprezentantem skupiny idiopatických intersticiálních pneumonií (IIP). Etiologie a patogeneze IPF není zatím plně objasněna, ale předpokládá se, že příčinou fibroprodukce, jako odpovědi na neznámý inzult, může být nerovnováha imunitní odpovědi s převahou Th2 cytokinů. Do roku 2011 nebyla dostupná žádná prokazatelně účinná léčba této závažné nemoci. Od března 2011, kdy byl k použití v Evropské unii schválen pirfenidon, má alespoň část nemocných s IPF šanci na potenciálně účinnou farmakologickou léčbu. V pokročilých fázích nemoci však pacientům zbývá jako jediná šance na prodloužení přežití a zlepšení kvality života transplantace plic. Pokud nejsou vhodnými kandidáty této léčebné metody, pak jim může být poskytnuta pouze léčba symptomatická.
Úvod
IPF je velmi závažné onemocnění, které postihuje difuzně obě plíce. Předpokládaná střední doba přežití je při této diagnóze 2,5 až 3 roky. Podkladem IPF je patologické hojení alveolárních lézí s nadprodukcí vazivové tkáně. Doporučení pro diagnostiku a léčbu této závažné nemoci se v poslední době velmi dynamicky mění na podkladě lepšího poznávání imunopatogeneze této nemoci a na podkladě klinických studií s novými léky, které jsou přímo zacíleny na patogenetické mechanismy IPF. Naopak opouštěny jsou léta zavedené léčebné postupy, u kterých nebyl prokázán účinek na prodloužení života, snížení mortality, zpomalení poklesu plicních funkcí a zlepšení kvality života. Současná diagnostická a léčebná doporučení odrážejí závěry konsenzu světových respiračních společností z roku 2011 a výsledky relevantních klinických hodnocení [1, 2].
Definice IPF
Poslední definice z roku 2011 popisuje IPF jako specifickou formu chronického fibrotizujícího intersticiálního procesu nejasné etiologie, který se objevuje u dospělých a je spojen s histologickým obrazem obvyklé intersticiální pneumonie (UIP) [1].
Epidemiologie
IPF postihuje asi 5 milionů lidí na celém světě. Prevalence této nemoci je celosvětově odhadována na 13–20/100 000 obyvatel a incidence na 6,8–16,3/100 000 obyvatel. Může však být i vyšší, protože lze předpokládat poddiagnostikovanost této choroby. IPF se vyskytuje o něco častěji u mužů než u žen. Incidence onemocnění stoupá s věkem. Pacienti s IPF jsou nejčastěji středního věku, v rozmezí od 40 do 70 let. Přibližně dvě třetiny pacientů jsou starší 60 let. IPF nemá dle provedených výzkumů žádnou jistou geografickou distribuci. Vyskytuje se celosvětově se stejnou prevalencí, bez rozdílu ve městech i na vesnicích a bez jakékoliv asociace s rasou nebo příslušností k etniku. IPF se obvykle vyskytuje sporadicky, familiární případy jsou vzácné.
Doposud byla objevena řada faktorů s pravděpodobným vlivem na vznik a vývoj IPF. V kontrolovaných studiích bylo prokázáno, že kouření je potenciálním rizikovým faktorem. Pravděpodobnost vzniku onemocnění stoupá s narůstajícím počtem let kouření. Jako rizikové pro vznik IPF byly popsány také expozice různým organickým a anorganickým prachům, nicméně my se domníváme, že se u těchto pacientů mohlo jednat spíše o chronické exogenní alergické alveolitidy či pneumokoniózy, které mohou mít v pokročilém stadiu podobný klinický a radiologický obraz jako IPF. Na spuštění patogenetického procesu IPF může mít vliv také řada virových a atypických patogenů. U pacientů s IPF byla zjištěna vyšší incidence EBV (viru Epsteina a Barrové), chřipkového viru, CMV (cytomegaloviru), viru hepatitidy C, viru parainfluenzy, viru HIV-1 (human immunodeficiency virus-1), viru spalniček, HHV-6 (lidského herpesviru 6) a bakterií rodu Mycoplasma. Můžeme tedy usuzovat, že obecně jakýkoli infekční, nejspíše virový inzult může u disponovaného jedince nastartovat proces patologického hojení plicní tkáně.
Etiopatogeneze
Etiopatogeneze IPF není zatím zcela jasná, zdá se však, že se jedná o uniformní patologickou odpověď plicní tkáně na různá infekční i neinfekční agens. Dříve se předpokládalo, že IPF vzniká jako reakce na zánětlivý proces, jenž přešel do chronického stadia. Podle posledních studií se ale soudí, že IPF vzniká pravděpodobně v nezánětlivém terénu jako odpověď na neznámý stimul, který způsobuje opakované poškození výstelky plicních sklípků vyúsťující v nekontrolovatelné a progredující jizvení. Zánětlivá reakce se někdy může vyskytnout až sekundárně. Alveolární makrofágy jsou u IPF prostřednictvím Th2 cytokinů pravděpopodobně tzv. alternativně aktivovány, čímž pak zvyšují produkci fibronektinu a indukují tím fibrogenezi ve fibroblastech.
Přirozený průběh nemoci je u jednotlivých pacientů obtížně klinicky mapovatelný, neboť není možné klinické a radiologické nálezy v čase doložit opakovanými nálezy histologickými. Nicméně, dle typického vzhledu radiologického obrazu na počítačové tomografii (CT) hrudníku s vysokou rozlišující schopností (HRCT) je patrné, že IPF je časově značně heterogenní proces, s okrsky normální plicní tkáně, okrsky s aktivní fibrotizací a s okrsky konečného stadia fibrózy s obrazem voštinovité plíce. Tradiční pohled na průběh IPF je založen na představě o pomalém poklesu plicních funkcí vedoucím k respiračnímu selhání a smrti. Pravděpodobněji však vypadá teorie o mnohočetných inzultech způsobujících tzv. akutní exacerbace nemoci s rychlejším poklesem plicních funkcí [3].
Patogenetický proces IPF od alveolární léze až po fibrózu
Mnohočetné léze alveolární výstelky
Na začátku patogenetického procesu pravděpodobně dochází k mnohočetnému poškození alveolárního epitelu, kdy je obnažena bazální membrána a spuštěna kaskáda produkce cytokinů, chemokinů a enzymů, které se pak podílejí na dalším rozvoji a udržování procesu fibroprodukce a jizvení plicní tkáně. Dalším krokem pak je nábor („recruitment“) fibroblastů a myofibroblastů s další depozicí extracelulární matrix a progrese k nevratné fibróze alveolu.
Porucha oxidačně-redukčních dějů
Oxidativní stres se v patogenezi IPF uplatňuje hlavně při vzniku iniciálních mikroskopických lézí alveolárního epitelu, které jsou pak objektem patologického hojení s vystupňovanou fibroproliferací. Pravděpodobnou roli hraje nerovnováha mezi redukčními a oxidačními systémy v plicích, které zahrnují celou škálu mechanismů kontroly tvorby a odbourávání reaktivních kyslíkových radikálů (ROS).
Nerovnováha proteolytických enzymů
Nerovnováha systému proteáz/antiproteáz může být jednou z příčin udržování bludného kruhu nekontrolovatelné fibroproliferace. Hlavními reprezentanty proteázového/antiproteázového systému jsou enzymy matrixové metaloproteinázy (MMP) a tkáňové inhibitory matrixových metaloproteináz (TIMP). Z MMP se na patogenezi IPF podílejí zejména MMP-2, MMP-7 a MMP-9. Z TIMP se v patogenezi IPF účastní TIMP-1, TIMP-2, TIMP-3 a TIMP-4.
Nerovnováha cytokinového spektra
Prozánětlivé cytokiny
Tumor nekrotizující faktor alfa (TNF-α, tumor necrosis factor α) spolu s interleukinem 1 (IL-1) hrají roli spíše v iniciálních fázích poškození plicního parenchymu než u pokročilého onemocnění.
Cytokiny typu Th2
Hlavními cytokiny, které určují vzorec patologického hojení plicní tkáně směrem k fibróze, jsou IL-4 a IL-13. Oba tyto cytokiny podporují růst fibroblastů a produkci kolagenu a zároveň zvyšují produkci transformujícího růstového faktoru beta 2 (TGF-β2, transforming growth factor β2) lidskými bronchiálními epiteliemi.
Cytokiny typu Th1
Interferon gama (IFN-γ) v experimentu dokáže tlumit vývoj bleomycinem indukované fibrózy u myší cestou regulace produkce TGF-β1. U IPF je jeho regulační role potlačena, neboť alternativní aktivace makrofágů zvyšuje sekreci IL-13 a nikoli IFN-γ. IL-12 je naopak cytokinem působícím antifibroticky cestou indukce IFN-γ.
Profibrogenní cytokiny
Jedním z cytokinů, který hraje klíčovou roli v indukci fibrogeneze, je TGF-β. Indukuje transkripci kolagenu typu I a fibronektinu ve fibroblastech. V endotelu navíc reguluje produkci a aktivitu růstového faktoru fibroblastů FGF-2 (fibroblast growth factor), který má výrazný mitogenní vliv na pneumocyty II. typu. To může mít za následek poruchu fyziologické reepitelizace drobných alveolárních lézí s následným vznikem prefibrotických okrsků.
Porucha angiogeneze
Patogenetický proces u IPF je komplexní a zahrnuje nejen plicní parenchym, ale i vaskulaturu. Za normálních podmínek jsou v plicích angiogenní a angiostatické stimuly v rovnováze. V případě IPF však dochází k posunu této rovnováhy a k iniciaci aberantní angiogeneze. Hlavními faktory ovlivňujícími angiogenezi ve smyslu jejího zvýšení jsou bazický fibroblastový růstový faktor (bFGF), vaskulární endoteliální růstový faktor (VEGF) a angiogenní CXC chemokiny.
Vliv osy renin-angiotenzin (RAS)
Aktivace RAS se velmi pravděpodobně účastní v patogenezi plicní fibrózy a plicní hypertenze, a to svým účinkem vazokonstrikčním, proliferačním a profibrogenním.
Původ fibroblastů a jejich „nesmrtelnost“
Existuje několik teorií o původu fibroblastů a myofibroblastů, které jsou přítomny v plicní tkáni u pacientů s IPF. Jedna z nich předpokládá, že jde o rezidentní kmenové buňky, které se aktivují a proliferují v odpovědˇ na poranění. Je možný i jejich původ z buněk alveolárního epitelu, které projdou takzvanou epitelo-mezenchymální transdiferenciací (EMT). Některé teorie a studie však spíše podporují hypotézu o mimoplicním původu fibroblastů, které pocházejí z kostní dřeně, chovají se jako mezenchymální kmenové buňky a jsou chemotakticky atrahovány do míst tkáňového poranění, kde přispívají k fibrotizaci plicní tkáně. Je pravděpodobné, že fibroblasty u jedinců s IPF mají i sníženou schopnost apoptózy a jsou chráněny proti apoptóze mediované Fas receptory. Tuto ochranu zprostředkovávají antiapoptotické faktory ze skupiny inhibitorů apoptózy (IAP).
Klinický obraz
IPF se klinicky projevuje progredující námahovou a posléze klidovou dušností, snadnou unavitelností, kašlem a v pozdějších fázích při nastupující hypoxemii i cyanózou. I když je pro IPF typický pozvolný a plíživý nástup dušnosti a její pozvolná progrese, u některých pacientů se vyskytnou epizody tzv. akutní exacerbace IPF. Tehdy dojde k náhlému klinickému zhoršení s poklesem plicních funkcí a radiologickým obrazem tzv. mléčného skla, který svědčí pro probíhající floridní rozsáhlé alveolární poškození s následnou rapidní progresí fibrózy. Akutní exacerbace, zvláště u pacientů s pokročilou IPF, jsou téměř bez výjimky u všech nemocných smrtelné.
Fyzikální nález
U tří čtvrtin pacientů se vyskytují fenotypové projevy, jako jsou paličkovité prsty s nehty tvaru hodinového sklíčka a poslechový fenomén krepitu (křoupání) slyšitelný nad plicními bazemi.
Funkční vyšetření plic
Typickou funkční poruchou je u pacientů s IPF restrikční ventilační porucha (RVP) s poruchou difuzní kapacity a snížením plicní poddajnosti. Vzácně se u pacientů s IPF setkáme i s obstrukční ventilační poruchou (OVP), a to u podskupiny nemocných s kombinací plicní rozedmy a plicní fibrózy. Difuzní kapacita je významně snížena, a to ve větší míře, než by odpovídalo redukci celkové plicní kapacity (TLC).
Pro IPF je typické výrazné zhoršení dušnosti již při submaximální zátěži a sníženy jsou vrcholová spotřeba kyslíku a ventilační rezerva. Při vyšetření krevních plynů je u pacientů s IPF v úvodu onemocnění patrný pokles saturace krve kyslíkem (Sa02) a pokles parciálního tlaku kyslíku (Pa02) v tepenné krvi pouze při zátěži, a teprve s progresí onemocnění dochází i ke klidové hypoxemii. Na základě hodnot krevních plynů je pak zvažováno u pacientů s IPF suplementární podávání kyslíku, generovaného buď koncentrátorem kyslíku, nebo aplikovaného ze zásobníku kapalného kyslíku. Dobrou informaci o funkčním stavu pacienta nám poskytne šestiminutový test chůzí (6-MWT), který je také nedílnou součástí vyšetření pro případnou indikaci přidělení kapalného kyslíku.
Zobrazovací metody
Skiagram hrudníku
V časném stadiu IPF může být skiagram hrudníku normální, případně se zjišťuje jemná oboustranná retikulace, zvláště v dolních plicních polích a v axilárních částech plic. S postupujícím onemocněním se zvýrazňuje retikulace až retikulonodulace s cystami o velikosti 2–20 mm a s obrazem voštinovité plíce. Současně dochází i ke zmenšení objemu plic s vyšším uložením bránice.
CT hrudníku s vysokou rozlišovací schopností
HRCT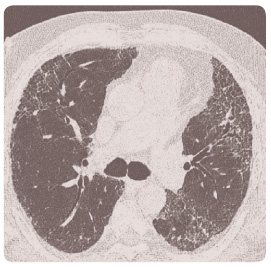 je klíčovým vyšetřením pro stanovení diagnózy a určení prognózy IPF. HRCT je schopno identifikovat IPF již v časných stadiích, kdy je onemocnění potenciálně léčitelné. IPF v HRCT obraze odpovídá UIP. Ta je charakterizována subpleurální a bazální retikulací s tlustostěnnými cystami různé velikosti a voštinovitou plicí, která se objevuje u více než 90 % nemocných. Častým nálezem jsou trakční bronchiektazie, popřípadě i bronchioloektazie. Vyznačena je obvykle distorze plicní architektoniky (obr. 1) [4].
je klíčovým vyšetřením pro stanovení diagnózy a určení prognózy IPF. HRCT je schopno identifikovat IPF již v časných stadiích, kdy je onemocnění potenciálně léčitelné. IPF v HRCT obraze odpovídá UIP. Ta je charakterizována subpleurální a bazální retikulací s tlustostěnnými cystami různé velikosti a voštinovitou plicí, která se objevuje u více než 90 % nemocných. Častým nálezem jsou trakční bronchiektazie, popřípadě i bronchioloektazie. Vyznačena je obvykle distorze plicní architektoniky (obr. 1) [4].
Bronchoalveolární laváž
Bronchoalveolární laváž je vyšetření, které má v případě IPF zejména význam z hlediska diferenciální diagnózy. Technicky se provádí fibrobronchoskopem, který zaklíňujeme do bronchu, a to většinou do bronchu pro střední lalok (v případě maxima změn v jiné oblasti můžeme volit i jiný bronchus), a pak vyplachujeme přilehlou plicní tkáň vlažným fyziologickým roztokem (obvykle aplikujeme 4 frakce po 50 ml). Získaný materiál je pak zpracováván pro cytologické vyšetření ultracentrifugací, pro cytometrické a mikrobiologické vyšetření tekutinu necentrifugujeme. Pro IPF je typické v cytologickém nálezu zmnožení granulocytů obvykle s malou příměsí eozinofilů, počet lymfocytů bývá zvýšen minimálně.
Plicní biopsie
Obvykle je pro diagnózu IPF dostačující syntéza klinického obrazu a HRCT nálezu kompatibilního s IPF obvyklého typu. Pokud máme o diagnóze pochybnosti, kupříkladu při netypickém HRCT obraze, je vhodné indikovat chirurgickou, obvykle videothorakoskopickou, plicní biopsii.
Histopatologický obraz
H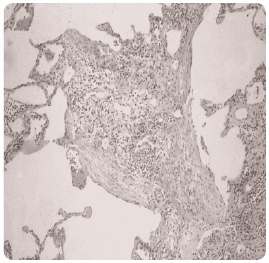 istopatologický obraz IPF odpovídá UIP se základními rysy: ložiskové postižení plicního parenchymu, fibrotické změny nestejného stáří („časová heterogenita“ procesu), fibroblastické fokusy a cysty obklopené pruhy husté fibrotizace (voštinovitá plíce), viz obr. 2.
istopatologický obraz IPF odpovídá UIP se základními rysy: ložiskové postižení plicního parenchymu, fibrotické změny nestejného stáří („časová heterogenita“ procesu), fibroblastické fokusy a cysty obklopené pruhy husté fibrotizace (voštinovitá plíce), viz obr. 2.
Diagnóza
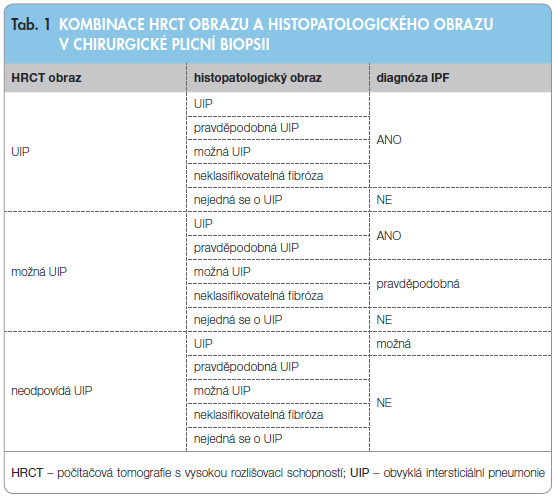
Diagnostická kritéria dle nového konsenzu světových respiračních společností z roku 2011 značně zjednodušují přístup k diagnostice IPF. Pokud je klinický a radiologický nález typický a jsou vyloučeny systémové nemoci pojiva a exogenní příčiny onemocnění plicního intersticia, pak není chirurgická biopsie nutná [1, 5].
Diagnostická kritéria IPF
- Vyloučení jiných příčin intersticiálních plicních procesů (domácí a profesní expozice, systémové nemoci pojiva, léková toxicita).
- Přítomnost HRCT vzorce UIP u pacientů bez plicní biopsie.
- Specifické kombinace HRCT a histopatologického (chirurgická plicní biopsie) UIP vzorce u pacientů s plicní biopsií (tab. 1).
Diferenciální diagnóza
 V rámci diferenciální diagnostiky je nejobtížnější odlišení fibrotické nespecifické intersticiální pneumonitidy (NSIP), chronické exogenní alergické alveolitidy, sarkoidózy IV. stadia a plicní fibrózy typu UIP v rámci systémových onemocnění. Při stanovení diagnózy v případě systémových onemocnění pojiva nám obvykle pomůže přítomnost klinických známek mimoplicního postižení a laboratorní průkaz autoprotilátek. K odlišení chronické exogenní alergické alveolitidy (EAA) je důležitá anamnéza expozice, případně pozitivita specifických IgG protilátek proti podezřelému antigenu. V ostatních případech se obvykle neobejdeme bez plicní biopsie. Pro odlišení sarkoidózy IV. stadia by mohla stačit biopsie transbronchiální, jinak je indikována biopsie chirurgická, obvykle videothorakoskopická.
V rámci diferenciální diagnostiky je nejobtížnější odlišení fibrotické nespecifické intersticiální pneumonitidy (NSIP), chronické exogenní alergické alveolitidy, sarkoidózy IV. stadia a plicní fibrózy typu UIP v rámci systémových onemocnění. Při stanovení diagnózy v případě systémových onemocnění pojiva nám obvykle pomůže přítomnost klinických známek mimoplicního postižení a laboratorní průkaz autoprotilátek. K odlišení chronické exogenní alergické alveolitidy (EAA) je důležitá anamnéza expozice, případně pozitivita specifických IgG protilátek proti podezřelému antigenu. V ostatních případech se obvykle neobejdeme bez plicní biopsie. Pro odlišení sarkoidózy IV. stadia by mohla stačit biopsie transbronchiální, jinak je indikována biopsie chirurgická, obvykle videothorakoskopická.
Léčba
Při tvorbě nového konsenzu pro diagnostiku a léčbu IPF byly provedeny metaanalýzy validních studií z posledních deseti let a vyhodnoceny léčebné postupy užívané pro léčbu IPF do roku 2009 [1]. Potvrdil se očekávaný závěr, že u IPF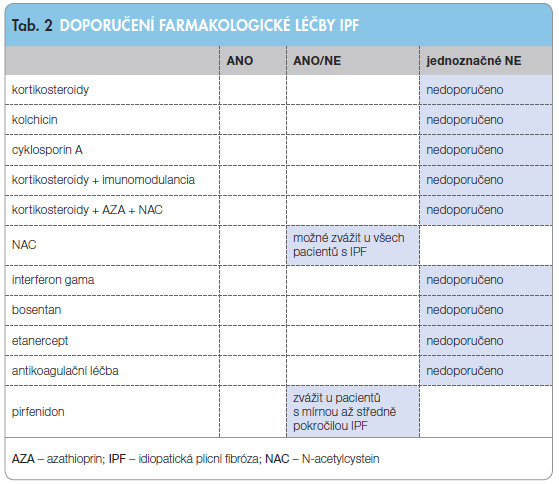 nejsou prakticky vůbec účinné protizánětlivě působící léky, jako jsou kortikosteroidy, imunosupresiva a cytostatika, a tudíž by se neměly nadále používat v léčbě IPF, a to ani samotné, ani v kombinacích, viz tab. 2. Tento závěr byl ještě podpořen interim analýzami studie Panther zveřejněnými na jaře 2012, na základě kterých bylo pozastaveno pokračování studijní větve pacientů s IPF léčných kombinací prednison + azathioprin + N-acetylcystein (NAC), neboť zde byla zjištěna vyšší mortalita oproti větvi se samotným NAC i ve srovnání s větví, v níž bylo podáváno placebo [6, 7].
nejsou prakticky vůbec účinné protizánětlivě působící léky, jako jsou kortikosteroidy, imunosupresiva a cytostatika, a tudíž by se neměly nadále používat v léčbě IPF, a to ani samotné, ani v kombinacích, viz tab. 2. Tento závěr byl ještě podpořen interim analýzami studie Panther zveřejněnými na jaře 2012, na základě kterých bylo pozastaveno pokračování studijní větve pacientů s IPF léčných kombinací prednison + azathioprin + N-acetylcystein (NAC), neboť zde byla zjištěna vyšší mortalita oproti větvi se samotným NAC i ve srovnání s větví, v níž bylo podáváno placebo [6, 7].
Pirfenidon je prvním lékem, který zasahuje přímo do patogeneze IPF. Inhibuje fibrózu indukovanou TGF-β1-blokádou nukleární translokace proteinů SMAD. Efektivita léčby pirfenidonem, a to optimálně v denní dávce 2403 mg, je patrná u pacientů s mírnou až středně těžkou IPF, bohužel u pacientů s pokročilým onemocněním nebyl takový efekt prokázán. Z nežádoucích účinků jsou nejmarkantnější fotosenzitivita a anorexie, jinak je léčba pacienty dobře tolerována.
V ČR je lék dostupný pro pacienty s mírnou až středně pokročilou IPF již 12 měsíců, zprvu v rámci programu na jméno pacienta a nyní cestou schválení úhrady mimořádně dovezeného registrovaného léku [8]. Lék by měl být podáván do progrese onemocnění. V případě jeho podávání je nutné mít na paměti potenciální hepatotoxicitu léku a v pravidelných intervalech monitorovat hodnoty jaterních testů. Výrazná je i kožní fotosenzibilita až fototoxicita, pacienti se proto musí při léčbě chránit před osluněním, a to pokud možno zakrytím většiny těla oděvem a ošetřením exponované části kůže ochrannými krémy s vysokým faktorem (SPF 50). Z nezávažných, přesto ale obtěžujících nežádoucích účinků jsou v popředí zažívací obtíže, především nauzea, nechutenství, případně průjmy. Obvykle se dají zvládnout podáváním léku spolu s jídlem, někdy je však zapotřebí snížit dávku na nejvyšší tolerovanou.
U všech pacientů s IPF bychom měli indikovat léčbu NAC v dávce 3krát 600 mg denně s cílem zabránit dalšímu poškozování alveolárního epitelu oxidačními ději a tím bránit tzv. akutním exacerbacím. NAC podáváme u pacientů trvale, jde o dobře tolerovatelný lék, některým pacientům vadí pouze zvýšená expektorace. Adherenci pacientů k této léčbě významně zhoršuje fakt, že NAC byl zcela vyňat z úhrad z prostředků veřejného zdravotního pojištění, a je tedy pro řadu pacientů obtížně finančně dostupný.
Z důvodu prevence akutní exacerbace IPF podáváme všem pacientům také inhibitory protonové pumpy v běžných denních dávkách, neboť bezpříznakový extraezofageální reflux může být také příčinou nových alveolárních lézí, a proto i spouštěčem akutních exacerbací [1].
Akutní exacerbace
Akutní exacerbace jsou závažnou epizodou v průběhu IPF. Vznikají na podkladě rozsáhlého alveolárního poškození s následnou rychle progredující fibrózou s poklesem plicních funkcí a respiračním selháním. Doposud zkoušené léčebné postupy se bohužel míjejí účinkem a většina pacientů na akutní exacerbaci IPF umírá. Nicméně při exacerbaci podáváme pacientům vysoké dávky kortikosteroidů parenterálně (obvykle 0,5–1 g methylprednisolonu i.v. po dobu 3 dnů s následným snížením na 1 mg/kg i.v.) a při podezření na současnou infekci jako spouštěč podáváme také antibiotika.
Při respirační insuficienci v průběhu akutní exacerbace ústící až v respirační selhání se nekloníme k umělé plicní ventilaci, neboť pro pacienty většinou není žádným přínosem, dochází při ní k ještě rozsáhlejšímu poškození plicní tkáně s následným úmrtím pacienta. Umělou plicní ventilaci bychom tedy volili pouze u těch pacientů s akutní exacerbací, kteří jsou již zařazeni na čekací listinu transplantace plic, a mají tedy alespoň nějakou šanci se darované plíce dočkat.
Dušnost u pacientů v terminálních stadiích IPF obvykle tlumíme opiáty, a to většinou v náplasťových formách s pomalým uvolňováním.
V případě pokročilého onemocnění s hypoxemií indikujeme pacientům, kteří splní kritéria České pneumologické a ftizeologické společnosti, dlouhodobou domácí oxygenoterapii, a to buď koncentrátorem kyslíku, nebo kyslíkem kapalným, který je indikován pro pacienty vyžadující vysoké průtoky kyslíku a pro pacienty mobilní, kdy umožňuje rehabilitaci i pohyb mimo domácí prostředí s předplněnou lahví kapalného kyslíku. Pro některé přísně selektované pacienty je vhodná transplantace plic, a to většinou plíce jedné, vzácně bloková transplantace srdce a plic. Kritérii pro zařazení na čekací listinu jsou v době diagnózy: dušnost, TL CO (transfer factor of the lung for carbon monooxide) < 40 % náležitých hodnot, desaturace < 88 % PaO2 a HRCT s obrazem voštinovité plíce. V případě longitudinálního sledování je indikací ke zvážení transplantace plic pokles usilovné vitální kapacity (FVC) o 10 % a pokles TL CO o 15 % spolu se zhoršením fibrózy na HRCT hrudníku.
Rehabilitace
Jednou z možností, jak zlepšit kvalitu života pacienta, je zlepšení jeho funkční výkonnosti a zmírnění dušnosti. Rehabilitace musí být v případě IPF komplexní, zahrnující učení, poradu a behaviorální techniky ke zlepšení sebeobsluhy, dále redukci symptomů a optimalizaci funkční kapacity. Probíhají studie zaměřené na efekt rehabilitace u pacientů s IPF a zdají se být slibné. Ve zkoumaných souborech bylo aplikováno cvičení na zlepšení svalové síly a trénink na bicyklovém ergometru. Po čtyřech týdnech s frekvencí cvičení 2krát týdně po 30 minutách došlo ke zmírnění dušnosti a zlepšení kvality života pacientů. V další studii byl rehabilitační trénink osmitýdenní s následným zlepšením výsledků testu s rumpálem, shuttle walk testu, zmírněním dušnosti a zlepšením kvality života. Zdá se tedy, že komplexní rehabilitace je vhodná i pro pacienty s IPF, kteří jsou výrazně fyzicky limitovaní dušností [9].
Monitorace klinického průběhu onemocnění a sledování efektu léčby
K monitorování klinického průběhu je doporučováno vyšetření plicních funkcí, ostatní vyšetřovací modality nejsou doporučeny. Jako progrese onemocnění je definován pokles FVC o 10 % absolutních hodnot a pokles TL CO o 15 % absolutních hodnot. Interval sledování je doporučen 3–6měsíční. Pokud však máme podezření na tzv. akutní exacerbaci IPF, je na prvním místě provedení HRCT hrudníku, kde najdeme obraz mlhovitých opacit superponovaný na obraz fibrózy. V případě podezření na infekci či nádor v terénu IPF jsou další vyšetřovací postupy včetně radiologického vyšetření samozřejmostí.
Prognóza
Prognóza je obecně špatná, navzdory léčbě má IPF v porovnání s ostatními typy idiopatických intersticiálních pneumonií téměř vždy progredující průběh, více než polovina pacientů umírá do 2–3 let od stanovení diagnózy. Prognóza je horší u pacientů s pokročilejším postižením plic dle HRCT vyšetření a s horšími či rychleji klesajícími plicními funkcemi. Většina studií hodnotících přežití u pacientů s IPF byla ale prováděna ještě v době před zavedením pirfenidonu do léčby IPF. Můžeme tedy předpokládat, že by se přežití u pacientů s IPF mohlo pod vlivem této léčby prodlužovat. U IPF je bohužel také ve zvýšené míře pozorován bronchogenní karcinom a ischemická choroba srdeční a žilní trombóza.
Zkratky IPF
AZA – azathioprin; bFGF – bazický fibroblastový růstový faktor; CMV – cytomegalovirus; EAA – exogenní alergická alveolitida; EBV –virus Epsteina a Barrové; EMT – epitelo-mezenchymální transdiferenciace; FGF – růstový faktor fibroblastů; FVC – usilovná vitální kapacita; HHV – lidský herpesvirus; HRCT – počítačová tomografie s vysokou rozlišovací schopností; IAP – inhibitory apoptózy;IIP – idiopatická intersticiální pneumonie; IL – interleukin; INF – interferon; IPF – idiopatická plicní fibróza; MMP – matrixová metaloproteináza; NAC – N-acetylcystein; NSIP – nespecifická intersticiální pneumonitida; OVP – obstrukční ventilační porucha; RAS – renin-angiotenzinový systém; ROS – reaktivní kyslíkové radikály; RVP – restrikční ventilační porucha; TGF – transformující růstový faktor; TIMP – tkáňové inhibitory matrixových metaloproteináz; TLC – celková plicní kapacita; TL CO – transfer factor of the
lung for carbon monooxide; TNF – tumor nekrotizující faktor; UIP – obvyklá intersticiální pneumonie; VEGF – vaskulární endoteliální
růstový faktor; 6-MWT – šestiminutový test chůzí
Seznam použité literatury
- [1] Raghu G, et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic Pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183: 788–824.
- [2] Du Bois RM. Strategies for treating idiopathic pulmonary fibrosis. Nat Rev Drug Discov 2010; 9: 129–140.
- [3] Strieter RM, Mehrad B. New mechanisms of pulmonary fibrosis. Chest 2009; 136: 1364–1370.
- [4] Fishbein MC. Diagnosis: to biopsy or not to biopsy: assessing the role of surgical lung biopsy in the diagnosis of idiopathic pulmonary fibrosis. Chest 2005; 128: 520S–525S.
- [5] Flaherty KR, King TE, Raghu G, et al. Idiopathic interstitial pneumonia. What is the effect of a multidisciplinary approach to diagnosis? Am J Respir Crit Care Med 2004; 170: 904–910.
- [6] Wells AU, Behr J, Costabel U, et al. Triple therapy in idiopathic pulmonary fibrosis: an alarming press release. Eur Respir J 2012; 39: 805–806.
- [7] The Idiopathic Pulmonary Fibrosis Clinical Research Network. Prednisone, Azathioprine and N-Acetylcysteine for Pulmonary Fibrosis. N Engl J Med 2012; 366: 1968–1977.
- [8] Azuma A, Taguchi Y, Ogura T, et al. Pirfenidone Clinical Study Group in Japan. Exploratory analysis of a phase III trial of pirfenidone identifies a subpopulation of patients with idiopathic pulmonary fibrosis as benefiting from treatment. Respir Res 2011; 12: 143. Published online 2011 October 28. doi: 10.1186/1465-9921-12-143.
- [9] Swigris JJ, Brown KK, Make BJ, et al. Pulmonary rehabilitation in idiopathic pulmonary fibrosis: A call for continued investigation. Resp Med 2008; 102: 1675–1680.