Lékové a léčebné interakce při antiarytmické terapii
Lékové interakce mohou být příčinou toxicity a nežádoucích účinků léčiv nebo naopak příčinou snížení jejich účinnosti. Podle mechanismu vzniku rozdělujeme lékové interakce na farmaceutické, farmakodynamické a farmakokinetické. Farmakokinetické lékové interakce spojené s biotransformací léčiva mohou nastat například na úrovni metabolismu zprostředkovaného enzymatickým systémem cytochromu P-450. Rozdílné reakce jednotlivých pacientů na stejnou farmakoterapii mohou být podmíněny genetickým polymorfismem, který vede k odlišnostem v aktivitě enzymů, receptorů a transportních mechanismů. V léčbě arytmií stojí v popředí riziko proarytmie, zejména v podobě výskytu komorových tachyarytmií typu torsades de pointes. V budoucnosti bude možno přesněji individualizovat farmakoterapii dle analýzy genomu. Zatím se však musíme soustředit na dodržování určitých klinických zásad bezpečného vedení léčby.
Úvod
Pojem léková interakce označuje situaci, kdy při současném podání dvou nebo více léčiv dojde ke změně účinku některého z nich. Může se jednat o účinek synergický, antagonický nebo kvalitativně změněný [1, 2]. Lékové interakce mohou být příčinou toxicity a nežádoucích účinků léčiv nebo naopak příčinou snížení či vymizení dříve stabilizovaného terapeutického účinku. Podle mechanismu vzniku rozdělujeme lékové interakce na farmaceutické, farmakodynamické a farmakokinetické [1].
Farmaceutické interakce představují spíše farmaceutickou inkompatibilitu, která je většinou známa předem a nutí nás k dodržování správných postupů při aplikaci léčiv a výběru vhodného vehikula či nosného infuzního roztoku při nitrožilní aplikaci léků apod. Farmakodynamické interakce probíhají často na úrovni buněčných receptorů. Farmakokinetické interakce mohou nastat při absorpci, distribuci, biotransformaci a exkreci léčiv [1].
Problematika lékových interakcí se týká samozřejmě i farmakologické léčby arytmií. Situace je zde v určitém ohledu rizikovější, neboť výsledkem některých interakcí je proarytmie, což je souhrnný název pro zhoršení manifestace arytmií či výskyt nového typu arytmie v průběhu aplikace různých léčiv. Obávaným jevem je komorová proarytmie typu torsades de pointes (TdP), která může být i fatální.
Během léčby arytmií navíc můžeme pozorovat i vzájemné interakce farmakologické a nefarmakologické léčby. Ty zahrnují například ovlivnění detekce arytmie a změnu stimulačního či defibrilačního prahu implantovaných přístrojů vlivem podávaných léčiv. Následující text je věnován některým hlediskům lékových a léčebných interakcí při ovlivnění arytmií.
Mechanismus proarytmického efektu léčiv
Různá léčiva mohou ovlivňovat tvorbu i šíření vzruchu v srdeční tkáni. Obávaným nežádoucím účinkem léčiv je jejich vliv na repolarizační fázi srdečního cyklu, což se v případě ovlivnění komorového myokardu manifestuje prodloužením QT intervalu a vznikem závažných komorových tachyarytmií typu TdP, které mohou způsobit zástavu oběhu a mohou přejít i v terminální fibrilaci komor. Arytmogenní mechanismus je v tomto případě podmíněn inhibicí draslíkových kanálů Iks a Ikr, která způsobí prodloužení repolarizační fáze akčního potenciálu s uplatněním membránových oscilací během fáze III charakteru tzv. časné následné depolarizace (EAD – early afterdepolarization). Při dosažení určité prahové hodnoty může EAD spustit komorovou extrasystolu až komorovou tachykardii (obr. 1).
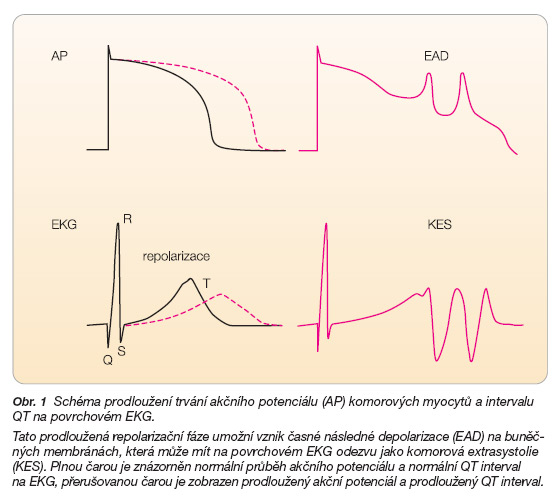
Dalším faktorem, který přispívá ke vzniku arytmií, je disperze průběhu repolarizace v komorovém myokardu. Délka akčního potenciálu se liší v subendokardiálních, subepikardiálních a „midmyokardiálních" myocytech (obr. 2).
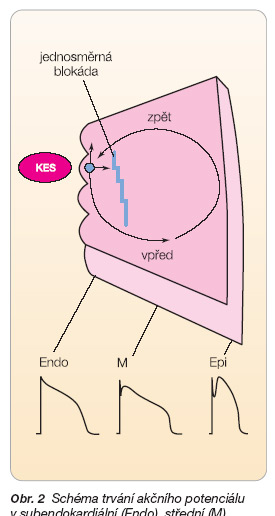
Je známo, že buňky ve střední vrstvě komorového myokardu (M-buňky) mají výrazně delší trvání akčního potenciálu v důsledku prodloužené repolarizace [3]. Vlivem některých léčiv se tyto rozdíly v trvání akčních potenciálů v různých vrstvách komorového myokardu mohou zvýraznit a tato disperze refrakterností a délky repolarizace přispívá ke vzniku arytmogenního reentry mechanismu (obr. 3).
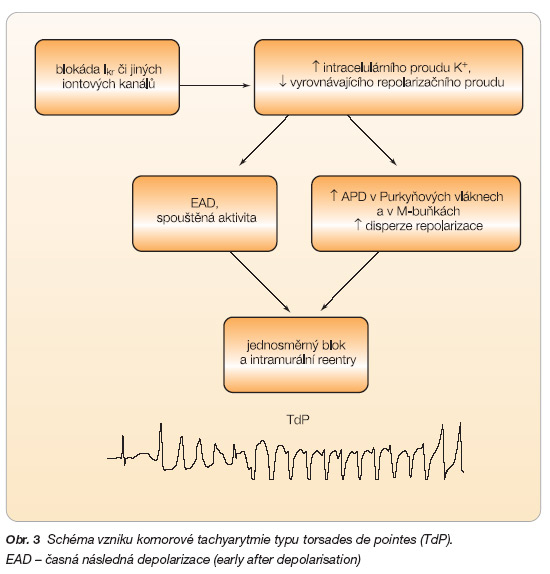
Vzhledem k tomu, že manifestace arytmogenních mechanismů může nastat vlivem farmak a zejména lékových interakcí, pokusíme se shrnout vybrané základní poznatky o daném problému.
Lékové interakce a riziko proarytmie
Farmakodynamické interakce jsou do určité míry předvídatelné, pokud máme dostatečné informace o farmakodynamických vlastnostech jednotlivých léčiv. Interakce lze očekávat, pokud současně podávaná léčiva účinkují na stejné receptory, na stejný orgán nebo na stejné regulační mechanismy. Tyto účinky mohou být – jak je již uvedeno výše – synergické nebo antagonické.
Farmakokinetické interakce vedou ke změně plazmatických (a následně tkáňových) koncentrací a mohou mít mnoho variací [2]. Mohou nastat během absorpce vlivem změny pH v gastrointestinálním traktu, vlivem skladby potravy, při změnách střevní motility, změnách střevní bakteriální flóry či při změnách průtoku krve v mezenteriální oblasti. Interakce léčiv s potravou jsou zatím jen málo prozkoumané. Obecně platí, že lipofilní látky jsou lépe vstřebány, pokud jsou podávány při jídle. Biologická dostupnost jednotlivých léčiv kolísá mimo jiné dle časového vztahu k příjmu potravy a jejího složení.
Interakce během biotransformace léčiv jsou nejčastější a nejzávažnější. Jde v podstatě o kompetici v enzymatické reakci. V případě inhibice je snížena metabolická eliminace dané látky, takže může dojít k předávkování. V případě indukce metabolismu je naopak snížena koncentrace léčiva, což může způsobit jeho poddávkování.
Farmakokinetické lékové interakce při biotransformaci léčiva mohou být například na úrovni metabolismu zprostředkovaného cytochromovým enzymatickým systémem.
Klíčovou roli zde hraje cytochrom P-450 a jeho jednotlivé izoformy. Izoenzymy cytochromu P-450 jsou zodpovědné za většinu přeměn cizorodých látek (včetně léčiv) v organismu [4]. Vyskytují se v játrech, v tenkém střevě, plicích, ledvinách a v menší míře i v dalších orgánech. V lidském genomu bylo nalezeno několik desítek izoenzymů cytochromu P-450 (zkráceně CYP), na metabolizaci léčiv se však významněji podílí jen několik z nich (nejrozšířenější CYP3A4, dále CYP2D6 a další). Jejich označení vyplývá z rozdělení tzv. superrodiny enzymů CYP do rodin, podrodin a na jednotlivé enzymy. Toto dělení uvedených enzymů respektuje jejich aminokyselinovou strukturu. Nebezpečí lékových interakcí vyplývá jak z možné indukce uvedených enzymů, tak z faktu, že při současném podání dvou či více látek, které jsou substrátem pro určitý enzym, dochází mezi těmito látkami ke kompetici o příslušný enzym. Nejvíce pravděpodobné jsou farmakokinetické interakce na úrovni metabolismu zprostředkovaného již výše zmíněným cytochromovým systémem CYP3A4 a CYP2D6 [4, 5].
Izoenzym CYP3A4 je nejvýznamnější systém metabolismu léčiv: jeho substrátem jsou mimo jiné léky též antiarytmika. Jeho typickými inhibitory jsou amiodaron, některá antimykotika či makrolidová antibiotika a grapefruitová šťáva. Typickými induktory uvedeného enzymového systému jsou dexamethason, barbituráty, rifampicin a třezalka. Stručný přehled substrátů, inhibitorů a induktorů CYP3A4 je uveden v tab. 1.

Zvýšení hladiny CYP3A4 (vlivem induktorů) je podstatou jednoho z typů farmakokinetických interakcí na úrovni metabolismu zprostředkovaného izoenzymem CYP3A4, jehož důsledkem je snížení hladiny metabolizovaného léčiva a vzrůst hladiny metabolitu [4]. Dalším častým typem lékových interakcí v rámci metabolismu cestou CYP3A4 je soutěž dvou či více léčiv podléhajících přeměnám na CYP3A4 o vazebné místo na tomto enzymu. To způsobí zvýšení hladiny „znevýhodněného" léčiva. Tab. 1 shrnuje jen velmi neúplný seznam substrátů pro CYP3A4, přesto je z tohoto přehledu zřejmé, jak různorodá léčiva kompetují o uvedený enzymový systém.
Dalším prvkem ve hře, který je zodpovědný za rezistenci k léčbě, je proteinový přenašeč – glykoprotein P. Je to ATP-dependentní pumpa, která kontroluje vstřebávání a vylučování některých léčiv (například digoxinu). Vzhledem k překrývání substrátové specifity mezi CYP3A4 a P-glykoproteinem a vzhledem k podobnostem induktorů a inhibitorů CYP3A4 a P-glykoproteinu je mnoho lékových interakcí společných pro CYP3A4 a P-glykoprotein [4, 6].
Enzymový systém CYP2D6, na rozdíl od CYP3A4, není inducibilní; zatím nejsou známy žádné látky, které by zvyšovaly jeho hladiny. Variabilita aktivit CYP2D6 ve vzorcích jaterní tkáně u lidí je vysvětlována genetickým polymorfismem. Ten podmiňuje tři hlavní fenotypy: pomalí metabolizátoři (s defektními CYP2D6 alelami), rychlí metabolizátoři (s „wild type" alelou) a ultrarychlí metabolizátoři s amplifikovanými geny pro funkční enzym CYP2D6 [5]. Dle literárních údajů je v kavkazské populaci zhruba 7 % defektních genů, vedoucích k fenotypu pomalého metabolizátora, zatímco v asijské populaci je frekvence defektních alel podstatně vyšší (téměř 50 %).
Účinek snížené aktivity CYP2D6 na průběh léčby má zajímavé klinické důsledky. Například vysoké hladiny antiarytmik mohou být příčinou proarytmického efektu, zvýšené hladiny tricyklických antidepresiv mohou mít kardiotoxické účinky.
Typickým substrátem pro enzym CYP2D6 jsou b-blokátory a tricyklická antidepresiva, typickými inhibitory CYP2D6 jsou například amiodaron, chinidin, celekoxib, cimetidin, ranitidin a kokain (tab. 2).

Nežádoucí lékové interakce mohou nastat například při současném podávání b-blokátorů s některými antidepresivy. Například fluoxetin (inhibitor zpětného vychytávání serotoninu) i jeho aktivní metabolit jsou substrátem pro CYP2D6. Při podávání tohoto antidepresiva pacientovi, který současně dostával metoprolol, byla zaznamenána závažná bradykardie v důsledku sníženého metabolismu (a tedy výrazně zvýšené hladiny) metoprololu [7].
Obdobné nálezy byly zaznamenány u kombinace karvedilolu či propranololu s fluoxetinem [8, 9].
Také nesteroidní antirevmatikum celekoxib zpomaluje inhibicí CYP2D6 metabolizaci metoprololu a může přispět k manifestaci vedlejších účinků léčby uvedeným b-blokátorem [10].
Chinidin je substrátem pro CYP3A4 a je zároveň účinným inhibitorem CYP2D6.
Je známo, že i subterapeutické dávky chinidinu inhibují výrazně CYP2D6, což může podstatně zvýšit plazmatickou hladinu propafenonu, pokud by byla podávána kombinace obou uvedených antiarytmik.
Amiodaron inhibuje všechny cytochromové systémy a také glykoprotein P. Amiodaron je metabolizován převážně CYP3A4 a tato skutečnost dává možnost lékových interakcí se substráty a inhibitory CYP3A4. U sotalolu nehrozí nebezpečí interakcí na úrovni enzymů CYP, neboť toto antiarytmikum nepodléhá biotransformaci s účastí cytochromu P-450 [4].
Indikace, vedení terapie a posuzování účinnosti antiarytmické medikamentózní léčby představuje poměrně složitý klinický problém. Je třeba si uvědomit, že i při podávání jednoho antiarytmického léčiva existuje interindividuální variabilita metabolismu (rychlí a pomalí metabolizátoři, rychlí a pomalí acetylátoři). Je to podmíněno genetickým polymorfismem enzymů zúčastněných na obou hlavních fázích metabolismu léčiv. Ve fázi I dochází k modifikaci molekuly látky (oxidace, redukce, acetylace, hydrolýza aj.), dochází ke zvýšení hydrofility molekuly, a tím k jejímu snadnějšímu vyloučení ledvinami. Ve fázi II dochází ke konjugaci látek nebo jejich metabolitů s kyselinou glukuronovou a sírovou a tento proces vede k inaktivaci léčiv a rovněž usnadňuje jejich následné vyloučení z organismu.
Vlivem některých látek může dojít k indukci (zmnožení) enzymů, což vede ke zrychlení biotransformace určité látky, a tak ke snížení její koncentrace v plazmě, a tím ke snížení jejího účinku.
Indukcí a inhibicí biotransformačních enzymů lze vysvětlit mnohé interakce současně podávaných léčiv. Rizikovější situace nastane, pokud pacient navíc trpí renální nebo hepatální insuficiencí, která může zpomalit obvyklý proces eliminace léčiva.
Další faktory zvyšující riziko proarytmie
I když byly již dříve publikovány kazuistiky upozorňující na riziko zhoršení arytmie po léčbě antiarytmiky, teprve po publikaci studie CAST (Cardiac Arrhythmia Suppression Trial) začala být věnována odpovídající pozornost této problematice – tj. proarytmickému efektu léčby antiarytmiky. Je známo, že též velké množství různých jiných léčiv (tj. nikoliv antiarytmik) může působit proarytmicky. Jsou to převážně látky prodlužující trvání akčního potenciálu [11, 12].
Je možno uvést některá antibiotika, antihistaminika, antimalarika, prokinetika, steroidy atd. [13–17] (tab. 3).

Je známa řada faktorů, které mohou riziko proarytmického efektu léčiv zvýšit. Především se jedná o abnormální průběh repolarizace v důsledku změněné funkce některých iontových kanálů na membránách srdečních myocytů. Výrazně vyššímu riziku lékové proarytmie jsou vystaveni především pacienti s kongenitální formou syndromu dlouhého QT (LQTS – long QT syndrome). Tito pacienti však představují jen malou část rizikové populace. Podstatně častější je situace u latentní formy LQTS, respektive tzv. snížené repolarizační rezervy [13–15]. Některé další faktory zvyšující riziko proarytmie – zejména v podobě komorové tachyarytmie typu TdP – shrnuje tab. 4. Při zahajování a vedení antiarytmické léčby je třeba věnovat pozornost všem uvedeným faktorům, neboť riziko proarytmie nemusí být vždy podmíněno čistě lékovou interakcí.

Léčebné interakce v terapii arytmií
Dle různých údajů až 50 % pacientů, kteří mají implantované přístroje (zejména implantabilní kardioverter-defibrilátor, ICD) je současně léčeno antiarytmiky [18, 19]. Tento fakt může přinášet další klinické problémy vyplývající z interakce uvedených léčebných postupů.
Jak již bylo naznačeno výše, různá antiarytmika mohou ovlivnit defibrilační práh, stimulační práh, délku cyklu komorové tachykardie a průběh komorového elektrogramu a mohou tak znesnadnit detekci tachykardie a mohou též ovlivnit průběh odpovědi na antitachykardickou stimulaci [19]. Rovněž provedení transtorakální elektrické kardioverze může ovlivnit další snímání intrakardiálních signálů a ovlinit též stimulační práh. Další zajímavou oblastí je modifikace arytmogenního substrátu katetrizační ablací či revaskularizací a z toho vyplývající modifikace dalšího výskytu a průběhu arytmií a jejich ovlivnění implantovaným systémem. Problematika těchto léčebných interakcí však přesahuje rámec tohoto sdělení a zmiňuji ji proto jen okrajově.
Závěr
Při lékových interakcích jde o modifikaci reakce na receptorech a mediátorech, na proteinových nosičích a na enzymech metabolizujících léčiva. Současné metodiky již umožňují některé změny reakcí identifikovat včas a rozpoznat hrozící riziko závažnějších nežádoucích účinků. V budoucnu lze očekávat, že analýza genomu podá informace o individuální variabilitě a perspektivně umožní individuální terapeutický přístup, který zaručí odpovídající účinnost a bezpečnost léčby jednotlivých pacientů [20]. Dříve než bude možno rutinně využívat tento přístup individuální farmakoterapie „šité na míru" jak výběrem léčiva, tak individualizací jeho dávkování, lze doporučit důsledné dodržování několika zásad, které sníží riziko nežádoucích lékových interakcí a zvýší bezpečnost medikamentózní léčby:
– ‑vyvarovat se polypragmazie,
– ‑brát v úvahu okolnosti zvýšeného rizika proarytmie (strukturální onemocnění srdce, hypokalémie, polymorbidita, vyšší věk, bradykardie, snížené hepatální a renální funkce),
– ‑monitorovat srdeční rytmus, QTc, minerály v séru – při používání léčiv ovlivňujících repolarizaci,
– ‑při očekávaném vyšším riziku proarytmie zahajovat léčbu při hospitalizaci (zajistit monitoraci EKG),
– ‑vyvarovat se používání kombinací antiarytmik,
– ‑zahajovat léčbu nižšími dávkami léčiva,
– ‑znát hlavní farmakokinetické charakteristiky léčiva, hodnotit efekt léčby až po odpovídajícím časovém intervalu (po dosažení „steady state"),
– ‑myslet na možnost proarytmie (i při chronické léčbě, kterou pacient dosud toleroval),
– ‑využít nefarmakologické léčebné postupy, hybridní léčbu arytmií (která nabízí vyšší léčebný potenciál a nižší riziko nežádoucích účinků).
Seznam použité literatury
- [1] Květina J, Grundmann M. Farmakologické interakce. Klin Farmakol Farm 2003; 1: 17–21.
- [2] Jerie P. Lékové interakce. Cor Vasa 2000; 42: 155–157.
- [3] Drouin F, Charpentier F, Gauthier C, et al. Electrophysiological characteristics of cells spanning the left ventricular wall of human heart: evidence for the presence of M-cells. J Am Coll Cardiol 1995; 26: 185–192.
- [4] Kousalová L, Baranová J, Anzenbacher P. Lékové interakce na úrovni cytochromů P-450 – Část I. Interakce na úrovni CYP3A4. Klin Farmakol Farm 2003; 17: 151–157.
- [5] Baranová J, Anzenbacher P, Kousalová L. Lékové interakce na úrovni cytochromů P-450 – Část II. Interakce na úrovni CYP2D6. Klin Farmakol Farm 2004; 18: 102–107.
- [6] Lin JH, Zamazali M. Role of P-glycoprotein in pharmacokinetics: clinical implications. Clin Pharmacokinet 2003; 1: 59–98.
- [7] Walley T, Pirmohamed M, Proudlove C, Maxwell D. Interaction of metoprolol and fluoxetine. Lancet 1993; 341: 967–968.
- [8] Graff DW, Williamson KM, Pieper JA, et al. Effect of fluoxetine on carvedilol pharmacokinetics, CYP2D6 activity, and autonomic balance in heart failure patiens. J Clin Pharmacol 2001; 41: 97–106.
- [9] Drake WM, Gordon GD. Heart block in a patient on propranolol and fluoxetine. Lancet 1994; 343: 425.
- [10] Werner U, Werner D, Rau T, et al. Celecoxib inhibits metabolism of cytochrome P450 2D6 substrate metoprolol in humans. Clin Pharmacol Ther 2003; 74: 130–137.
- [11] Haverkamp W, Breithardt G, Camm AJ, et al. The potential for QT prolongation and proarrhythmia by nonantiarrhythmic drugs: clinical and regulatory implications. Eur Heart J 2000; 21: 1216–1231.
- [12] Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev 2005; 85: 1205–1253.
- [13] Roden DM. Taking the „idio“ out from „idiosyncratic“: Predicting torsades de pointes. PACE 1998; 21: 1029–1034.
- [14] Napolitano C, Schwarz P, Brown AM, et al. Evidence for a cardiac ion channel mutation underlying drug-induced QT prolongation and life-threatening arrhythmias. J Cardiovasc Electrophysiol 2000; 11: 691–696.
- [15] Camm AJ, Malik M, Yap YG. Acquired long QT syndrome. Blackwell Futura, Oxford, UK, 2004; 60–86.
- [16] Albrecht CA. Proarrhythmia with non-antiarrhythmics. Cardiology 2004; 102: 122–139.
- [17] Boyer EW. Antiarrhythmic agents. In: Intensive Care Medicine. Ed. Irwin RS, Rippe JM. Lippincott Williams and Wilkins, Philadelphia, 2003; 1512–1524.
- [18] Thomas AC, Moser SA, Smutka ML, Wilson PA. Implantable defibrillation: Eight years clinical experience. PACE 1998; 11: 2053–2058.
- [19] Santini M, Pandozi C, Ricci R. Combining antiarrhythmic drugs and implantable devices therapy: benefits and outcome. J Interv Card Electrophysiol 2000; 4: 65–68.
- [20] Bultas J. Farmakogenetika aneb správný lék ve správné dávce pro správného nemocného. Remedia 2003; 13: 152–157.