Morbus Gaucher – možnosti diagnostiky a terapie
Gaucherova nemoc je dědičná porucha metabolismu s autozomálně recesivním typem dědičnosti, při které dochází k lyzozomálnímu střádání glukosylceramidu v buňkách makrofágového původu. Onemocnění je způsobeno poruchou enzymu b-glukocerebrosidázy, která štěpí glukocerebrosid na glukózu a ceramid. V genu pro b-glukocerebrosidázu, který se nachází na chromozomu 1q21, již bylo nalezeno více než 300 mutací. Podle klinického průběhu onemocnění se Gaucherova nemoc dělí na tři typy. Non-neuronopatický typ (bývalý typ I), který se projevuje progredující hepatosplenomegalií, bolestmi dlouhých kostí a známkami krvácivé diatézy. Akutní neuronopatický typ (bývalý typ II) se projevuje v kojeneckém věku neprospíváním, strabismem nebo nystagmem, progredující hepatosplenomegalií, postižením CNS se zástavou psychomotorického vývoje a opistotonem. Subakutní neuronopatický typ (bývalý typ III) začíná v batolecím nebo předškolním věku, má vystupňované viscerální symptomy, postihuje CNS, ale probíhá pomaleji než typ II. Diagnóza Gaucherovy nemoci se provádí enzymatickým vyšetřením aktivity b-glukocerebrosidázy v izolovaných leukocytech a molekulárním vyšetřením genu pro b-glukocerebrosidázu. Léčba je možná tzv. enzymatickou substituční terapií – nitrožilními infuzemi rekombinantního enzymu, nebo tzv. substrát redukční terapií.
Úvod
Gaucherova nemoc je autozomálně recesivně dědičné onemocnění, které řadíme k tzv. lyzozomálním střádavým onemocněním. Tato onemocnění jsou většinou způsobena nedostatečnou aktivitou některé z kyselých hydroláz, které jinak štěpí složité látky vznikající např. při degradaci buněčných stěn. V důsledku nedostatečné aktivity daného enzymu pak dochází k nahromadění substrátu nad enzymatickým blokem v lyzozomech. U Gaucherovy nemoci je snížena aktivita enzymu glukocerebrosidázy (β-glukosidázy), nedegradovaný substrát – glukocerebrosid (glukosylceramid) se hromadí v lyzozomech cirkulujících nebo fixních makrofágů. Glukocerebrosidáza je kódována genem nacházejícím se na 1. chromozomu (1q21), v současné době je známo cca 300 mutací tohoto genu. Onemocnění morbus Gaucher se vyskytuje s incidencí cca 1 : 60 000 panetnicky, výrazně častější incidence je v populaci tzv. aškenázských Židů, kde je uváděna četnost výskytu 1 : 500 až 1 : 1000.
Podle klinických projevů a průběhu onemocnění rozlišujeme tři základní typy:
-
Adultní, viscerální, chronický typ I – nejčastější typ onemocnění se začátkem v pozdním dětství až dospělosti. Projevuje se progredující splenomegalií, mírnou hepatomegalií, projevy krvácivé diatézy, bolestmi kostí, kloubů; progrese je pomalá, bez postižení CNS.
-
Akutní neuronopatický typ II (infantilní) – první projevy jsou zřetelné brzy po narození, většinou do šesti měsíců věku. Děti neprospívají, je patrný opistotonus, strabismus, masivní splenomegalie, může docházet k těžkým kožním změnám charakteru ichtyózy, psychomotorický vývoj je zastaven, k úmrtí dochází většinou do dvou let věku.
-
Subakutní neuronopatický typ III (juvenilní) – první projevy se objevují v časném dětství, kdy pozorujeme masivní hepatosplenomegalii, projevy krvácivé diatézy, kostní postižení i deformity páteře, od 1. dekády jsou patrné projevy postižení CNS – atypické pohyby hlavy a očí, epilepsie.
Historie
V roce 1882 bylo onemocnění poprvé popsáno lékařem Ernestem Phillipem Gaucherem jako významná splenomegalie nenádorového charakteru. V letech 1901–1912 bylo vypozorováno, že toto onemocnění postihuje více orgánových systémů současně a má familiární výskyt (Brill). V roce 1920 bylo poprvé identifikováno prudce probíhající neurologické postižení u infantilní formy, které bylo označeno jako II. typ morbus Gaucher (Kraus, Rusca). V roce 1934 byla identifikována látka, která se hromadí, a byla označena jako glukocerebrosid (Aghion). Typ dědičnosti byl popsán v roce 1950 (autozomálně recesivní dědičnost; Herndon a Bender). V roce 1958 zaznamenal Fried mnohonásobně vyšší incidenci tohoto onemocnění v židovské populaci Aškenázů a o rok později (1959) popsal Hillborg smíšený typ III se závažným viscerálním a pomalu progredujícím postižením CNS. V roce 1965 byl identifikován příslušný enzym – glukocerebrosidáza (Brady), od roku 1972 byla možná prenatální diagnostika (Schneider). V roce 1985 byla naklonována cDNA (Sorge, Tsuji) a popsána genetická heterogenita onemocnění. První klinický pokus o enzymatickou susbstituční terapii aglucerázou proběhl úspěšně v roce 1991 v USA (Barton, Brady), v České republice v roce 1995 (Zeman). Léčba byla zpočátku prováděna enzymem extrahovaným z placent, později byla nahrazena podáním rekombinantního enzymu imiglucerázy.
Klinické projevy u nejčastější formy morbus Gaucher (viscerální typ I)
Nahromadění glukocerebrosidu v kostní dřeni způsobuje útlak červené kostní dřeně a zhoršení podmínek pro krvetvorbu. Nejprve dochází k úbytku trombocytů, který se projevuje snadným vznikem modřin, častou epistaxí, protrahovaným krvácením po malých chirurgických či stomatologických výkonech, u dívek silnější menstruací. Později dochází k úbytku červených krvinek s typickými projevy anémie – bledostí a únavou. Někdy pozorujeme i leukopenii, neprojevuje se však zvýšená náchylnost k infekčním onemocněním. Expanze nastřádaného materiálu v kostní dřeni se kromě toho podílí i na útlaku krevního zásobení corticalis, může docházet k tzv. kostním infarktům, jejichž klinickým korelátem je kostní krize. Jedná se o velmi bolestivý stav, pacient je na několik dnů upoután na lůžko, klinické projevy mohou imitovat osteomyelitidu, často bývá přítomna i horečka a leukocytóza, hemokultura je však negativní. Dále můžeme pozorovat výskyt patologických fraktur dlouhých kostí, žeber, kompresivních fraktur obratlů, dochází k rozvoji tzv. Erlenmeyer flask deformity při remodelaci stehenních kostí, vyskytuje se osteopenie, často dochází k předčasným artrotickým změnám na velkých kloubech s nutností totální endoprotézy v poměrně mladém věku.
Dalším časným příznakem bývá výrazná splenomegalie, slezina často zasahuje až do malé pánve a přes střední čáru. Kromě nebezpečí poranění zvětšené sleziny nechráněné hrudním košem se mohou stupňovat příznaky cytopenie při projevech hypersplenismu. Zvětšená játra a slezina působí útlak žaludku, takže pacienti mají pocit nasycení i při velmi malém příjmu potravy. Zvětšené orgány v dutině břišní mohou omezovat i rozsah pohybů – obtížný předklon.
Celkově děti hůře prospívají, i když vzhledem k prominujícímu břichu při organomegalii není toto neprospívání na první pohled patrné. Je proto nutno sledovat antropometrické parametry, vývoj sekundárních pohlavních znaků a rozvoj puberty, který bývá u pacientů trpících Gaucherovou chorobou opožděn.
Diagnostika
Dle podrobné osobní či rodinné anamnézy je již mnohdy možné pomýšlet na diagnózu střádavého onemocnění či přímo na morbus Gaucher. Údaj o multisystémovém postižení progresivního charakteru je klíčový. Ve fyzikálním vyšetření bývá nápadná splenomegalie, později i zvětšení jater, mohou se objevit známky krvácivé diatézy na kůži či sliznicích. Typickými laboratorními nálezy jsou trombocytopenie, často trombocytopatie (porucha agregace), anémie, méně často leukopenie. V séru bývá patrná hyperimunoglobulinemie, vyšší hladina ferritinu, v imunoelektroforéze je možno pozorovat monoklonální gamapatii. Při vyšetření koagulací nacházíme obvykle prodloužení APTT (aktivovaný parciální tromboplastinový čas), eventuálně nižší hodnoty proteinu C a S, charakteristické jsou vysoké hodnoty D-dimerů.
Typickým nálezem pro střádavá onemocnění, hlavně pro Gaucherovu nemoc, jsou vysoké hodnoty tzv. biomarkerů. Patří k nim např. ACP (tartarát rezistentní kyselá fosfatáza – norma ≤ 100 nkat/l), dále chitotriosidáza (norma 4–195 nmol/mg prot./h) či protein CCL 18. Chitotriosidáza je protein, který bývá významně uvolňován aktivovanými makrofágy a u neléčených pacientů s morbus Gaucher dosahuje extrémně vysokých hodnot v řádech tisíců. Hladiny chitotriosidázy velice dobře korelují s aktivitou onemocnění, při podání adekvátní léčby rychle klesají. Bohužel asi 5 % populace je pro chitotriosidázu deficitních, proto nízké hodnoty chitotriosidázy při elevaci ACP či CCL18 proteinu střádání nevylučují.
Obraz střádavých změn ve dřeni dlouhých kostí či v tělech obratlů je možno prokázat pomocí MRI (magnetická rezonance) – snížení signálu v obrazech T1W i T2W. Při hodnocení nálezu je však nutno brát v úvahu také změny související s věkem, jako je postupná náhrada červené kostní dřeně tukovou žlutou kostní dření, snížení signálu je možno také mylně považovat za zánětlivé změny. Hodnocení proto musí vždy provádět zkušený radiolog, nejlépe se specializací na metabolické pacienty.
Typickým nálezem bývají střádavé buňky v bioptickém materiálu z kostní dřeně či z punkce sleziny – tzv. Gaucherova buňka má typický vzhled s cytoplazmou, která se podobá zmačkanému papíru – tzv. wrinkled paper appearance (obr. 1, poskytnuto společností Genzyme). Toto vyšetření indikujeme, objeví-li se v diferenciální diagnostice podezření na eventuální maligní proces. Jinak se především u dětí s typickou anamnézou, klinickými a laboratorními nálezy snažíme nejprve potvrdit diagnózu morbus Gaucher neinvazivními metodami. Jednoznačným potvrzením tohoto onemocnění je průkaz snížené aktivity β-glukosidázy (glukocerebrosidázy) v izolovaných leukocytech periferní krve či v kultuře fibroblastů. Molekulárně biologické vyšetření diagnózu verifikuje, někdy zároveň může vypovědět i o eventuálním dalším vývoji onemocnění. Např. 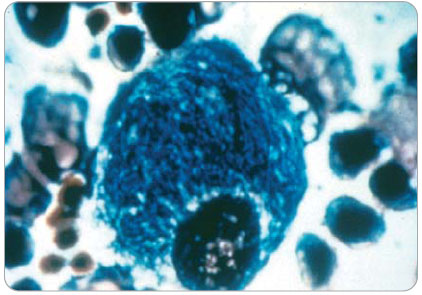 homozogyti pro mutaci L444P jsou pacienti s typicky těžkým průběhem onemocnění – subakutním neuronopatickým typem III. Naopak mutace N370S/N370S bývala spojována s lehkým průběhem bez neurologického postižení. Dnes již tento axiom neplatí, při delším dožívání pacientů s morbus Gaucher se velmi často právě u pacientů s touto mutací objevují příznaky parkinsonismu ve vyšším věku (5krát častěji než v běžné populaci). Obecně lze říci, že tíže onemocnění a rychlost progrese je nepřímo úměrná věku při počátku prvních projevů onemocnění. Tedy čím dříve se objeví první symptomy, tím rychlejší bude průběh, závažnější postižení a s tím související předpoklad zkrácené doby dožití.
homozogyti pro mutaci L444P jsou pacienti s typicky těžkým průběhem onemocnění – subakutním neuronopatickým typem III. Naopak mutace N370S/N370S bývala spojována s lehkým průběhem bez neurologického postižení. Dnes již tento axiom neplatí, při delším dožívání pacientů s morbus Gaucher se velmi často právě u pacientů s touto mutací objevují příznaky parkinsonismu ve vyšším věku (5krát častěji než v běžné populaci). Obecně lze říci, že tíže onemocnění a rychlost progrese je nepřímo úměrná věku při počátku prvních projevů onemocnění. Tedy čím dříve se objeví první symptomy, tím rychlejší bude průběh, závažnější postižení a s tím související předpoklad zkrácené doby dožití.
Terapie
Doporučenou léčbou je v první řadě tzv. enzymatická substituční terapie. V ČR je registrována imigluceráza (amp. s obsahem 400 a 200 jednotek), rekombinantní enzym podávaný nitrožilní infuzí v intervalu 2 týdnů. Doporučená dávka se pohybuje v rozmezí 30–60 j/kg/2 týdny, nutný je individuální přístup s úpravami dávkování dle klinické odezvy a laboratorních nálezů. Většinou je možno po 6–12 měsících dávku snížit na hodnoty v rozmezí 20–30 j/kg/2 týdny. Přibližně za 6 měsíců od zahájení této terapie se výrazně upravují až vymizí subjektivní obtíže, zlepší se hematologické parametry, zhruba do 12 měsíců pozorujeme postupný ústup organomegalie. Nálezy na kostech se upravují až s delším časovým odstupem – první známky regrese infiltrativních změn je možno sledovat většinou za 2–3 roky terapie. Výhodou této léčby je i možnost podání enzymu v průběhu gravidity a laktace. Nevýhodou je nutnost intravenózní aplikace, vysoká cena a nemožnost ovlivnit neurologické příznaky (enzym – velká molekula – nepřestupuje přes hematoencefalickou bariéru).
Další možností léčby je tzv. substrát redukční terapie. Pomocí miglustatu (n-butyl-deoxynojirimycin, cps.) je blokován enzym glukosyl transferáza, který jinak umožňuje vznik glukocerebrosidu z glukózy a ceramidu. Tím je redukováno celkové množství substrátu, a tudíž i stupeň střádání. Tato terapie funguje částečně na principu malých molekul s předpokládaným lepším průnikem do kostí a možností ovlivnění CNS. Částečně je popisován i efekt chaperonu – vazbou na aktivní místo vadného enzymu v endoplazmatickém retikulu opraví jeho terciární strukturu, čímž je enzym schopen transportu do lyzozomu, kde může uplatnit svoji katalytickou aktivitu. Zvyšuje tedy funkci vlastního reziduálního enzymu. Tato terapie je využívána pro pacienty ve stabilizovaném stavu, není vhodná k zahájení akutní terapie. Výhodou je tabletová forma (dávkování 3krát denně jedna tableta s obsahem 100 mg léčivé látky), nevýhodou je menší účinnost jak na ústup subjektivních obtíží, tak na normalizaci hematologických parametrů či organomegalie. Ovlivnění CNS či kostních příznaků je sporné, byla zvažována kombinace enzymové substituční terapie a substrát redukční terapie u neuronopatického typu III, v klinické studii však nebyl potvrzen jednoznačný efekt této kombinované terapie. Nevýhodou léčby miglustatem jsou především časté nežádoucí účinky – projevy nadýmání, nauzey, průjmy hlavně po požití potravin obsahujících laktózu. Při bezmléčné dietě se po několika týdnech stav upravuje a dietu je možno postupně uvolnit. U některých pacientů byl popsán rozvoj tremoru, poruch paměti či ztráty čití. Kvůli teratogennímu působení není léčba vhodná pro pacienty, kteří plánují založení rodiny.
Výhledově předpokládáme, že kromě enzymové substituční či substrát redukční terapie bude možno zvolit cílenou léčbu chaperony (tč. ve stadiu klinických studií) či genovou terapii. Součástí péče o pacienty je genetické poradenství, vyšetření sourozenců, partnerů před plánovaným rodičovstvím, eventuálně při určení diagnózy během gravidity prenatální diagnostika. Péče o pacienty se střádavým onemocnění by měla být komplexní, je nutno spolupracovat s celou řadou odborníků z různých oborů – kardiologie, respirační onemocnění, ortopedie, revmatologie, oční, ORL, kožní, psychologie, sociální poradenství.
Komplikace
U pacientů s neléčeným morbus Gaucher se častěji než v běžné populaci vyskytují nádorová onemocnění. Jednoznačně byl prokázán vyšší výskyt mnohočetného myelomu, plazmocytomu, hematologických malignit. Vyšší výskyt solidních tumorů nebyl jednoznačně potvrzen. Při hepatosplenomegalii dochází často k rozvoji portální hypertenze s vytvořením portokaválních anastomóz. Nebezpečné může být krvácení z jícnových varixů při současné trombocytopenii, trombocytopatii a koagulačních abnormitách. Ve vyšším věku byl u pacientů s Gaucherovou chorobou pozorován několikanásobně vyšší výskyt parkinsonismu, především u viscerálního typu I s mutací N370S v homozygotním i heterozygotním stavu. U neléčených pacientek byly popsány četné komplikace v průběhu gravidity – časté spontánní aborty či protrahované krvácení v časné poporodní době.
Diferenciální diagnostika
V diferenciálně diagnostické rozvaze je nutno pomýšlet především na maligní lymfom, leukemii s dřeňovým útlumem, při projevech krvácivé diatézy např. na hemofilii, autoprotilátkami podmíněnou či idiopatickou trombocytopenii, zvažovat jiné příčiny splenomegalie (infekce – CMV, EBV atd.), při postižení skeletu je nutno vyloučit kostní tumory, osteomyelitidu, při postižení kyčelního kloubu např. aseptickou nekrózu (Leg-Calvé-Perthes). Přítomnost protilátek proti trombocytům nevylučuje diagnózu morbus Gaucher – často pozitivní nález u prokázané diagnózy morbus Gaucher.
Situace v ČR
V ČR je v současné době sledováno 30 pacientů s prokázanou diagnózou morbus Gaucher, 29 pacientů trpí viscerálním typem I, 1 pacient je postižen subakutním neuronopatickým typem III. V souboru je 21 žen a 9 mužů, průměrný věk pacientů je 35 let (v rozmezí 12–58), průměrná doba léčení je 6 let (rozmezí 6 měsíců až 13 let), 2 pacienti jsou bez větších obtíží, dosud neléčeni, 5 pacientů užívá substrát redukční terapii miglustatem, 23 pacientů je léčeno substitucí enzymu – průměrná dávka imiglucerázy je 27 j/kg/2 týdny (rozmezí 15–35 j). Všichni pacienti jsou pravidelně sledováni – 1krát za 3–6 měsíců se provádí antropometrie, u dětí se sleduje PM vývoj, fyzikální vyšetření, základní laboratorní vyšetření včetně biomarkerů, 1krát ročně se provádějí zobrazovací metody – MRI páteře, dlouhých kostí, volumometrie jater a sleziny, sonografické vyšetření k posouzení eventuálních ložiskových změn v parenchymových orgánech či k posouzení nepřímých známek portální hypertenze, denzitometrie skeletu 1krát za 1–2 roky. U pacienta s typem III je vyšetření rozšířeno o neurologickou složku včetně EEG, vyšetření oční, ORL, spirometrické, evokovaných potenciálů a další, dle klinické symptomatologie.
Registr
Od roku 1997 existuje mezinárodní registr pro Gaucherovu nemoc, kde jsou shromažďována anamnestická data, fyzikální nálezy, doba stanovení diagnózy, projevy komplikací, průběh onemocnění, laboratorní data, výsledky zobrazovacích vyšetření, způsob terapie, případné nežádoucí reakce, popř. nutnost přerušení léčby či příčina úmrtí. V současné době je v registru evidováno více než 5000 pacientů z 61 zemí celého světa. Součástí registru jsou i údaje o všech pacientech z ČR, které jsme poskytli po získání informovaného souhlasu od našich pacientů.
Závěr
S pacienty, u nichž byla stanovena diagnóza morbus Gaucher, se můžeme setkat v ordinaci pediatra, hematologa, revmatologa či ortopeda. U multisystémového postižení s tendencí k progresi je proto nutné zvažovat možnost méně obvyklé diagnózy, střádavého onemocnění. Pomoci nám může důkladná anamnéza s výskytem typických obtíží u rodičů, prarodičů, sourozenců, v širší rodině. Cenným přínosem je i možnost vyšetření tzv. biomarkerů, které mohou napomoci v diferenciálně diagnostické rozvaze. Vzhledem k možnosti určení diagnózy pomocí neinvazivních metod (enzymologické vyšetření v izolovaných leukocytech periferní krve, molekulárněbiologické vyšetření) a vzhledem k možnosti účinné terapie je nutné na tuto diagnózu pomýšlet u pacientů s neobjasněnou trombocytopenií, splenomegalií, skeletálními obtížemi.
V současné době se objevují nové možnosti diagnostiky včetně cíleného vyšetření rizikové populace metodou tzv. suché kapky (zaschlá kapka krve na filtračním papírku, možno zaslat k enzymologickému vyšetření v laboratoři), diagnostikují se nová onemocnění, rozšiřují se terapeutické možnosti, u léčených pacientů se prodlužuje délka přežívání, mají možnost založit rodinu. Tyto jistě pozitivní skutečnosti však s sebou přinášejí celou řadu dalších otázek a problémů. Extrémní finanční náročnost terapie vzácných onemocnění neumožňuje poskytnout tuto terapii okamžitě všem diagnostikovaným pacientům. Je proto nutné diskutovat o určitých pravidlech, za jakých okolností tuto léčbu budeme zahajovat (indikační kritéria – léčba preklinická, či léčba až v případě rozvoje klinických obtíží), je-li možno terapii přerušit u jinak infaustně nemocných pacientů, u nespolupracujících pacientů, jak postupovat v případě, kdy matka odmítne možnost prenatální diagnostiky a narodí se dítě se střádavým onemocněním, atd. Problémy k diskusi se v této otázce objevují stále častěji a bude nutno k nim zaujmout určitá závazná stanoviska.
Kazuistika
Chlapec se narodil z první fyziologické gravidity s PH 4030 g a PD 51 cm. Poporodní adaptace i psychomotorický vývoj byly v normě. Při tříleté preventivní prohlídce byla zjištěna mírná hepatosplenomegalie. V 9 letech se nejdříve po fyzické zátěži a později i trvale objevily bolesti pravého kolenního kloubu. MR vyšetření ukázalo patologický signál dřeně v distální části pravého femuru, nález byl hodnocen jako pravděpodobný následek posttraumatických změn. V 11 letech byl pacient pro akutně vzniklé bolesti pravého kyčelního kloubu léčen antibiotiky s klinickou diagnózou coxitis l. dx., obtíže ustoupily. Ve 12 letech si chlapec při sportu zlomil levé předloktí, fraktura se zhojila bez komplikací. Krátce poté se opět po sportu objevily horečky, otok a bolestivost pravého kolenního kloubu s alterací celkového stavu. Pro podezření na akutní osteomyelitis byl pacient léčen antibiotiky (linkomycin) a byla provedena operační revize s implantací gentamicinu. Po přechodném zlepšení se znovu objevují septické teploty, progreduje splenomegalie, anémie (Hb 99 g/l, MCV 80 fl), leukopenie (4 x 109/l), trombocytopenie (103 x 109/l) a koagulopatie (APTT 47,1 s, kontrola 33; D-dimery > 1000, norma < 200). Při vyšetření kostní dřeně pro klinické podezření na hemoblastózu byly nalezeny pěnovité buňky svědčící nejspíše pro Gaucherovu nemoc (obr. 1).
Při metabolickém vyšetření ve věku 14 let dominovala bledost kůže a sliznic, mírná tachykardie a omezená hybnost pravé dolní končetiny po operační revizi. Chlapec měl výraznou hepatosplenomegalii (játra 5 cm pod 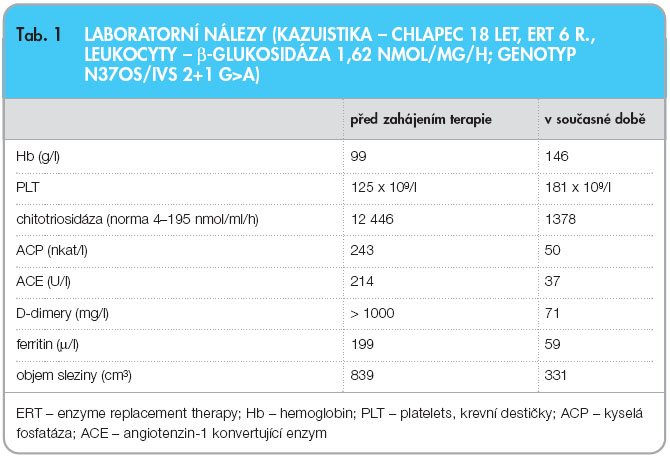 oblouk žeberní), slezina dosahovala k pupku. Mentální úroveň odpovídala věku. V laboratorním vyšetření byla nápadná vysoká aktivita kyselé fosfatázy (243 nkal/l, norma < 100) a chitotriosidázy (12 446 nmol/ml/h, norma < 190) svědčící pro aktivaci makrofágového systému a zvýšená hladina angiotenzin-1 konvertujícího enzymu (214 U/l, norma < 55). MR vyšetření zobrazilo infiltraci obratlových těl Th, L, S páteře i dřeně kostí dlouhých končetin. Denzitometrické vyšetření ukázalo snížený obsah kostního minerálu v bederní páteři (Z-skóre -2,3). Volumometrické vyšetření jater a sleziny pomocí MR potvrdilo mírnou hepatomegalii (objem 1419 cm3, norma pro věk < 800 cm3) a excesivní splenomegalii (839 cm3, norma < 80 cm3). Diagnózu Gaucherovy nemoci verifikovalo enzymatické vyšetření β-glukocerebrosidázy v izolovaných leukocytech (1,62 nmol/ml/hod, norma
oblouk žeberní), slezina dosahovala k pupku. Mentální úroveň odpovídala věku. V laboratorním vyšetření byla nápadná vysoká aktivita kyselé fosfatázy (243 nkal/l, norma < 100) a chitotriosidázy (12 446 nmol/ml/h, norma < 190) svědčící pro aktivaci makrofágového systému a zvýšená hladina angiotenzin-1 konvertujícího enzymu (214 U/l, norma < 55). MR vyšetření zobrazilo infiltraci obratlových těl Th, L, S páteře i dřeně kostí dlouhých končetin. Denzitometrické vyšetření ukázalo snížený obsah kostního minerálu v bederní páteři (Z-skóre -2,3). Volumometrické vyšetření jater a sleziny pomocí MR potvrdilo mírnou hepatomegalii (objem 1419 cm3, norma pro věk < 800 cm3) a excesivní splenomegalii (839 cm3, norma < 80 cm3). Diagnózu Gaucherovy nemoci verifikovalo enzymatické vyšetření β-glukocerebrosidázy v izolovaných leukocytech (1,62 nmol/ml/hod, norma 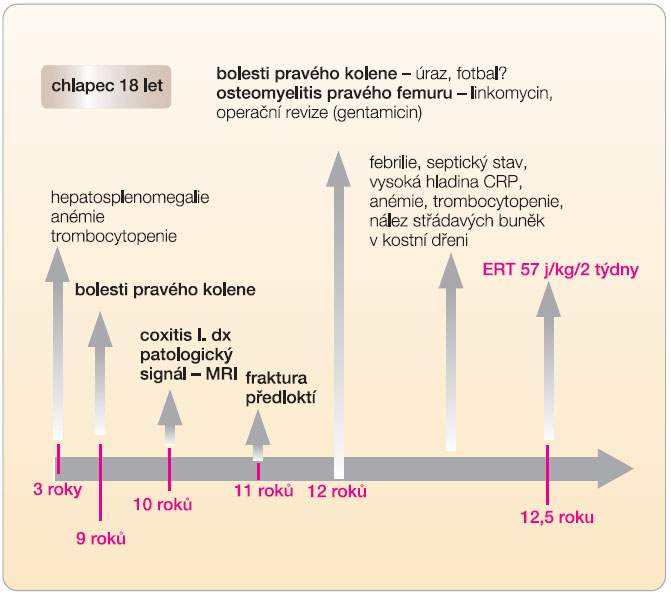 7,5–16) a DNA vyšetření, které ukázalo, že chlapec je složený heterozygot dvou mutací N370S/IVS2+1 G>A. Rodiče jsou zdraví heterozygoti. Shrnutí výsledků laboratorních testů viz tab. 1.
7,5–16) a DNA vyšetření, které ukázalo, že chlapec je složený heterozygot dvou mutací N370S/IVS2+1 G>A. Rodiče jsou zdraví heterozygoti. Shrnutí výsledků laboratorních testů viz tab. 1.
Po zhodnocení klinického stavu byla po dohodě se zdravotní pojišťovnou zahájena enzymatická substituční terapie rekombinantní β-glukosidázou (imigluceráza 60 j/kg 2krát měsíčně). Do půl roku vymizely bolesti kloubů a kostí a upravily se hematologické parametry. Po roce léčby dochází i k ústupu organomegalie. Kontrolní vyšetření MR po dvou letech dokumentuje ústup infiltrace kostní dřeně obratlů i dlouhých kostí, laboratorně trvá pouze mírná elevace chitotriosidázy. Vývoj obtíží u pacienta graficky znázorňuje obr. 2.
Seznam použité literatury
- [1] Pastores MG, Weinreb JN, Aerts H, et al. Advances in Gaucher disease: Therapeutics goals and evaluation and monitoring guidelines. Supplement to Seminars in Hematology Oct. 2004, Vol 41, No 4, Suppl 5.
- [2] Gaucher Registry. Annual Report 2009.
- [3] Mehta A. Epidemiology and natural history of Gaucher’s disease. Eur J Intern Med 2006; 17: 82–85.
- [4] Grabowski GA. Gaucher disease: gene frequencies and genotype/phenotype correlations. Genet Test 1997; 1: 5–12.
- [5] Pastores MG, Patel JM, Firooznia H. Bone and joint complications related to Gaucher disease. Curr Rheumatol Rep 2000; 2: 175–180.
- [6] Erikson A, Forsberg H, Nilsson M, et al. Ten years’experience of enzyme infusion therapy of Norrbottnian (type 3) Gaucher disease. Acta Paediatr 2006; 95: 312–317.
- [7] Hollak CE, Levi M, Berends F, et al. Coagulation abnormalities in type 1 Gaucher disease are due to low-grade activation and can be partly restored by enzyme supplementation therapy. Br J Haematol 1997; 96: 470–476.
- [8] Rosenbloom BE, Weinreb JN, Zimran A, et al. Gaucher disease and cancer incidence: a study from the Gaucher Registry. Blood 2005; 105: 4569–4572.
- [9] Zimran A, Ilan Y, Elstein D. Enzyme replacement therapy for mild patients with Gaucher disease. Am J Hematol 2009; 84: 202–204.
- [10] de Fost M, Out TA, de Wilde FA, et al. Immunoglobulin and free light chain abnormalities in Gaucher disease type I: data from an adult cohort of 63 patients and review of the literature. Ann Hematol 2008; 87: 439–449.
- [11] Boot RG, Verhoek M, de Fost M, et al. Marked elevation of the chemokine CCL18/PARC in Gaucher disease: a novel surrogate marker for assessing therapeutic intervention. Blood 2004; 103: 33–39.