Nealkoholická steatohepatitida a možnosti její léčby
Nealkoholická steatóza a nealkoholická steatohepatitida jater (NAFLD/NASH) je jaterní onemocnění charakterizované histologickým obrazem pozorovaným u alkoholické jaterní steatózy a steatohepatitidy u osob bez významné konzumace alkoholu (< 20 g ethanolu/den). Při prevalenci, která je odhadována na 20–30 % v široké populaci pro jaterní steatózu a na 2–3 % pro NASH, patří v industriálním světě k nejvýznamnějším jaterním onemocněním. NASH je pokročilou a progresivní fází NAFLD se vztahem k jaterní cirhóze a je potenciálně smrtelným onemocněním. Těsná souvislost s obezitou centrálního typu, diabetem 2. typu a hypertenzí umožňuje považovat NAFLD za jaterní komponentu metabolického syndromu. V etiopatogenezi NAFLD se uplatňuje akumulace tuků v periferii i v játrech vedoucí ke vzniku centrální i periferní inzulinové rezistence. Související mobilizace a porucha utilizace mastných kyselin vyvolává řadu pochodů, které vedou v hepatocytech k omezení -oxidace a ke snížení produkce energie, k oxidačnímu stresu, k další akumulaci triglyceridů. Vedou k poškozování jaterních buněk a k jejich zániku, k vyvolání zánětlivé reakce a ke vzniku fibrózy jaterní tkáně. V procesu přechodu od prosté steatózy ke steatohepatitidě a cirhóze i v prohlubování inzulinové rezistence se uplatňuje řada cytokinů, které mají původ v játrech i v tukové tkáni (TNF-, leptin, adiponektin, IL-6 a další). Zobrazovací metody umožní přesně kvantifikovat stupeň steatózy, k přesnějšímu posouzení přítomnosti a stupně zánětlivých a fibrózních změn je zapotřebí jaterní biopsie. V léčbě NAFLD/NASH se uplatňují především dietní a režimová opatření, která vedou ke snížení obsahu tuku v játrech i v periferii a k obnovení citlivosti tkání na inzulin. K redukci váhy lze přispět farmakologicky a rovněž metodami bariatrické chirurgie. Ve farmakoterapii NAFLD/NASH se uplatňují především látky zvyšující citlivost k inzulinu, zejména stimulátory PPAR- – glitazony. Ostatní léky – hypolipidemika, antioxidanty, hepatoprotektiva a probiotika – mají v léčbě NAFLD/NASH jen dílčí či žádný význam.
Key words: non-alcoholic hepatic steatosis – non-alcoholic steatohepatitis – PPAR-g activators – rosiglitazone.
Historie, definice a klasifikace NAFLD/NASH
Jen málokteré jaterní onemocnění vzbudilo v poslední době takovou pozornost jako nealkoholická steatohepatitida (NASH). Onemocnění bylo prvně popsáno Ludwigem v roce 1980 jako léze podobající se alkoholické jaterní steatohepatitidě u pacientů bez významné konzumace alkoholu [1]. Onemocnění do devadesátých let minulého století prakticky neznámé je nyní v celém industriálním světě považováno za nejčastější chronické a progresivní jaterní onemocnění se vztahem k jaterní cirhóze, jaternímu selhání i k hepatocelulárnímu karcinomu (tab. 1).
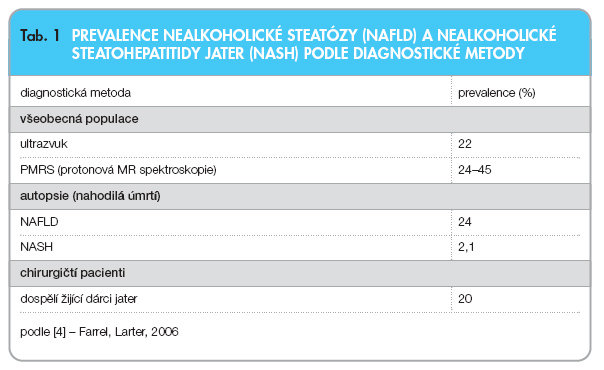
Na základě výskytu nemocných s nevysvětlitelnou elevací ALT ve Třetí národní studii zdraví a nutrice v USA (NHANES III) lze odhadovat prevalenci nealkoholické steatózy (NAFLD) v neselektované severoamerické populaci až na 7,3 % [2]. NAFLD/NASH je v současnosti nejčastějším důvodem návštěvy hepatologa ve Spojených státech, v Austrálii a na Novém Zélandu, je též četná v asijských zemích i v Evropě [3]. Prevalence v České republice není známa. Změny životního stylu, které vedou k nadměrnému kalorickému příjmu i k nevhodnému složení stravy a zároveň k poklesu fyzické aktivity, jsou považovány za klíčový faktor rostoucí incidence NASH. Pro konstantní vazbu NAFLD s rizikovými faktory aterosklerózy, jakými jsou DM 2. typu, obezita centrálního typu a hypertenze, je NAFLD často chápána jako jaterní komponenta metabolického syndromu (syndromu inzulinové rezistence) a tato těsná asociace ukazuje na podobné etiopatogenetické faktory [4].
Onemocnění je potenciálně závažné až letální. Přibližně u 9–25 % pacientů s NASH se vyvine jaterní cirhóza [3]. Asi 30–40 % pacientů s vyvinutou jaterní cirhózou v rámci NASH zemře na komplikace jaterního onemocnění do 10 let [5].
Definice NAFLD a NASH není přesně vymezená ani všeobecně přijatá. Podle konference AASLD věnované NASH, pořádané v roce 2002 v Atlantě (USA), je za NAFLD považována akumulace tuků v jaterních buňkách přesahující 5–10 % hmotnosti jater, a to u pacientů bez významné konzumace alkoholu, za kterou většina zúčastněných výzkumníků považovala 14 jednotek alkoholického nápoje za týden (≤ 20 g ethanolu/den). K histopatologické diagnóze NASH je krom přítomné steatózy zapotřebí nález lobulárního zánětu a perivenulární a portální fibrózy [6]. Na zmíněné konferenci byla přijata následující klasifikace NAFLD: třída 1– prostá steatóza, třída 2 – steatóza s lobulárním zánětem, třída 3 – regresivní (balonovité) změny hepatocytů, třída 4 – přítomnost Malloryho hyalinu nebo fibrózy. Fibróza při nálezu NAFLD je klasifikována do 4 stadií: 1. v zóně 3 – perivenulární, perisinusoidální nebo pericelulární fibróza, fokální nebo difuzní, 2. stejně jako ve stadiu 1, ale připojuje se ložisková nebo difuzní portální fibróza, 3. přemosťující fibróza ložisková nebo difuzní, 4. cirhóza s reziduální perisinusoidální fibrózou nebo bez ní [7]. NASH je tedy třeba považovat za pokročilou fázi NAFLD. Nutno podotknout, že symptomatická steatóza či steatohepatitida je přítomna nejen u alkoholické jaterní choroby, ale též u jiných dobře definovaných jaterních onemocnění, např. u Wilsonovy choroby, chronické hepatitidy C při infekci genotypem 3, u lékového poškození (amiodaron, tamoxifen, methotrexát a nukleosidová analoga), celiakie a abetalipoproteinémie a u dalších geneticky podmíněných onemocnění mitochondrií a mikrozomů. Symptomatická steatohepatitida při těchto chorobách do kategorie NAFLD nespadá.
Diagnostika NAFLD/NASH
K přesnému rozlišení NASH od benigní steatózy je zapotřebí jaterní biopsie. Přes snahy o zavedení a zpřesnění neinvazivních metod odhadu stupně fibrózy nelze její podíl a zejména přítomnost zánětlivé reakce ve steatotických játrech současnými metodami s dostatečnou přesností určit. Existuje obecná shoda, že podezření na prostou steatózu u obézního pacienta není třeba biopticky ověřovat. Klinické známky svědčící pro pokročilé fáze onemocnění se většinou bez biopsie neobejdou. Stejně tak pro odhad prognózy onemocnění se zdá být bioptické vyšetření jater nezbytné. V současnosti však přesně definovaná pravidla k indikaci bioptického vyšetření u nemocných s NAFLD/ NASH neexistují [6].
Klinické vyšetření
Základním klinickým nálezem u pacientů s NAFLD/NASH je hepatomegalie a laboratorní stanovení vyšší aktivity aminotransferáz ALT a AST v krvi. Sonografický průkaz „jasných jater" vede k diagnóze jaterní steatózy s vysokou pravděpodobností – senzitivita 89 % a specificita 93 % [8]. Tyto nálezy stačí při negativním výsledku screeningu nejběžnějších jaterních onemocnění vedoucích k symptomatické jaterní steatóze, a zejména při vyloučení možného podílu konzumace alkoholu > 20 g/den ke klinické diagnóze NAFLD. Vyšetření by mělo být doplněno o základní antropometrická měření (BMI, obvod pasu a boků). Většina populačních studií výskytu NAFLD/NASH je založena na výsledcích těchto vyšetření. Obtížnější je posoudit závažnost takto diagnostikované NAFLD, či lépe řečeno odlišit nekomplikovanou steatózu jater od závažné a progresivní NASH [9]. Zde je jistě nejpřesnějším nástrojem jaterní biopsie, bohužel v kontextu v současnosti celkem omezených léčebných implikací a vysokého podílu benigních nálezů je její provádění nepopulární a spíše výjimečné. Moderní zobrazovací metody poskytují přesnou kvantifikaci obsahu tuku v jaterní tkáni (CT, MR, eventuálně protonová spektrometrie a DEXA) [10]. Jejich specificita přesahuje hodnoty dosahované prostým ultrazvukovým vyšetřením, tyto metody však, stejně jako ultrasonografie, nejsou dostatečně senzitivní ani specifické pro stanovení stupně fibrózy jaterní tkáně, a již vůbec ne pro posouzení přítomnosti zánětlivých změn. Průkaz vaziva má pro diagnózu komplikované NAFLD zásadní význam. Pro širší diagnostické použití připadá v úvahu buď metoda FibroScan [11], nebo stanovení biochemických markerů fibrózy. Při vyšetřování fibrózy u NAFLD/NASH jsou publikovány zkušenosti se stanovením sérového hyaluronanu, aminoterminálního propeptidu kolagenu III a 7S domény kolagenu VI [12].
Etiopatogeneze vzniku NASH
NAFLD a NASH jsou považovány za jaterní komponenty metabolického syndromu a jejich vzájemný epidemiologický vztah je mimořádně těsný. Dle WHO je metabolický syndrom definován jako porušený metabolismus glukózy (diabetes nebo hyperglykémie nalačno nebo patologický glukózový toleranční test nebo průkaz inzulinové rezistence clampovou studií) při současné přítomnosti alespoň dvou následujících faktorů: poměr obvodu pasu a boků > 0,9 u muže a > 0,85 u ženy, hladina s-triglyceridů > 1,75 mmol/l nebo s-HDL cholesterolu < 0,9 mmol/l u muže a < 1,0 mmol/l u ženy, krevní tlak > 140/90 mm Hg a albuminurie > 20 mg/min (nebo poměr albumin/kreatinin > 30 mg/g). Pacienti s metabolickým syndromem mají přibližně dvojnásobnou pravděpodobnost mít vyšší aktivitu ALT (nevysvětlenou nálezem virové hepatitidy, hemochromatózy nebo zvýšenou konzumací alkoholu). Ze všech sledovaných faktorů metabolického syndromu byla s nevysvětlitelnou elevací ALT nejtěsněji svázána inzulinová rezistence (OR 2,64, 95% CI; 1,97–3,51) [13]. U pacientů s metabolickým syndromem je častější sonografický nález steatózy než ve zdravé populaci (OR 39, 96% CI 17,8–97) [14] a přibližně 40 % mužů a 26 % žen s metabolickým syndromem má sonograficky zjevnou steatózu [15].
Metabolické studie animální i humánní bez výjimky spojují viscerální adipozitu (obezitu centrálního typu), inzulinovou rezistenci a jaterní steatózu. Akumulace triglyceridů v jaterních buňkách je úzce svázána se vznikem inzulinové rezistence (obr. 1) [16].
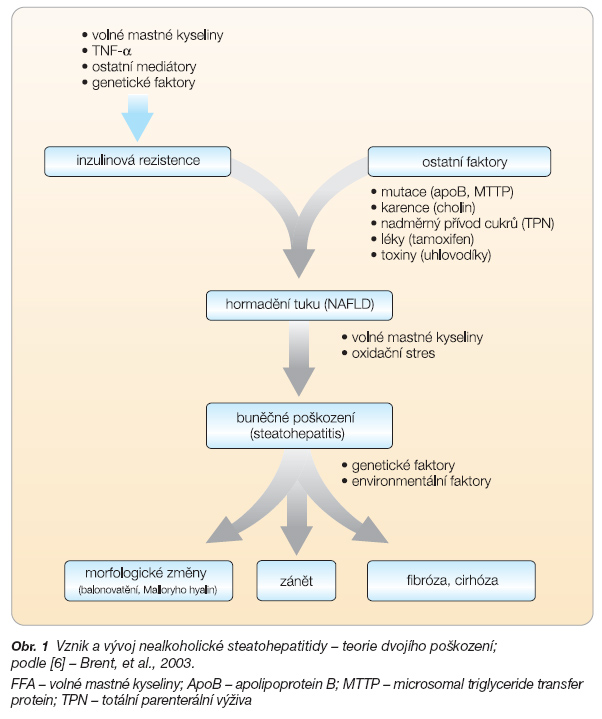
Inzulinová rezistence vede v periferních tkáních k lipolýze, mobilizaci volných mastných kyselin a k jejich vstupu do jater. Centrální inzulinová rezistence vede ke snížené produkci glykogenu v játrech, zvyšuje glukoneogenezi, omezuje b-oxidaci, a vede tak ke snížené utilizaci volných mastných kyselin a stimulaci de novo syntézy triglyceridů [17]. Tím dochází k další akumulaci triglyceridů v hepatocytech [18]. Steatóza hepatocytů prohlubuje inzulinovou rezistenci. Volné mastné kyseliny aktivují kinázy (též IKK-b), které chybně fosforylují IRS-1 a IRS-2. To vede k omezení stimulace PI-3 kinázy, která přenáší dál nitrobuněčný signál z inzulinu na glukózové přenašeče Glut-4, čímž je omezen příjem a utilizace glukózy hepatocyty i periferními tkáněmi [19]. V současnosti někteří autoři považují mitochondriální lézi za primární příčinu porušené oxidace mastných kyselin s následkem akumulace triglyceridů v jaterní tkáni, která vede ke vzniku centrální inzulinové rezistence [4, 20]. Podíl periferní inzulinové rezistence na vzniku NASH však zpochybněn není [21, 22]. Obnovení citlivosti tkání k inzulinu tak zůstává klíčovým faktorem v léčbě NAFLD.
Je známo několik dalších mechanismů, kterými volné mastné kyseliny (FFA) působí přímo či nepřímo hepatocelulární lézi. Volné mastné kyseliny indukují na povrchu hepatocytů expresi FAS molekul, jejichž interakce s jinými ligandami vedou k buněčné smrti [23]. Volné mastné kyseliny destabilizují lysozomy a vedou k uvolnění katepsinu B a k aktivaci NF-kB. NF-kB pak významně zvyšuje produkci TNF-a, cytokinu s klíčovým postavením v poškození jaterní tkáně, vzniku nekrózy hepatocytů, ale i v udržování stavu inzulinové rezistence [24]. Volné mastné kyseliny navíc inhibují aktivitu mitochondriálního dýchacího řetězce. Tento útlum vede ke generaci ROS a peroxidaci tuků, která způsobuje poškození mitochondriální DNA s dalšími důsledky pro jejich funkci. Jednou z možných kandidátních molekul inicializujících mitochondriální dysfunkci při NAFLD je již zmíněný TNF-a [25]. Mitochondrie reagují na oxidační poškození ze zvýšené produkce ROS indukcí ICP-2 (uncoupling protein 2), která ale vede nejen ke snížení produkce ROS, ale též ke snížení tvorby ATP a následnému riziku buněčné smrti hepatocytů nedostatkem energie [26]. Další metabolickou cestou zvyšující v hepatocytech oxidační stres, která byla popsána u metabolického syndromu, je zvýšená aktivita cytochromu CYP2E1. Vysoká aktivita je u metabolického syndromu spojována se zvýšenou koncentrací FFA a ketolátek. Zvýšená aktivace CYP2E1 přispívá rovněž ke zvyšování inzulinové rezistence, a to i bez přítomnosti steatózy [27]. Oxidační stres vznikající více mechanismy je v každém případě velmi významným faktorem vedoucím k progresi NAFLD do NASH.
Obezita je charakterizována zmnožením tukové tkáně, která je zdrojem adipokinů s převahou prozánětlivé aktivity. Převaha prozánětlivých a profibrogenních cytokinů vede k zánětu v jaterní tkáni (NASH) a následně i k její fibrotizaci a přestavbě. Akumulace tuku v hepatocytech vede k zánětlivé reakci. Zmiňované kinázy významné pro rozvoj inzulinové rezistence (IKK-b) aktivované přítomností FFA přispívají ke zvýšené produkci transkripčních faktorů (NF-kB). Ty pak vyvolávají expresi prozánětlivých cytokinů IL-1 a IL-6 [28] a tvorbu Cox-2, která podmiňuje syntézu dalších prozánětlivých mediátorů. Tukem indukovaná tvorba NF-kB aktivuje rovněž TNF-a, jehož koncentrace je ve steatotických játrech vždy zvýšena [29]. Není jasné, zda zvýšená koncentrace TNF-a přispívá více k prvotní akumulaci tuků mechanismem inzulinové rezistence, nebo zda je určující až pro rozvoj buněčného poškození.
Vznik fibrózy u pacientů s NAFLD je mediován jednak prozánětlivými cytokiny, jednak nezávisle na nich přímo adipokiny z tukové tkáně. Leptin, který se vyskytuje ve vysoké koncentraci u obezity i steatózy jater, aktivuje přímo stelární buňky [30], adiponektin naopak vede k inhibici jejich funkce a může vyvolat jejich apoptózu. Jeho koncentrace jsou však v situacích steatózy nízké [31]. Dalším z významných mediátorů jaterní fibrózy u nemocných s metabolickým syndromem je angiotenzin II, který zvyšuje proliferaci, migraci jaterních hvězdicovitých buněk a jejich produkci kolagenu [32].
Není zcela jednoznačné, které z výše uvedených mechanismů se uplatňují jako „prvotní poškození" jaterní tkáně při vzniku NAFLD a které vytvoří „second hit", klíčový pro vývoj NASH a jaterní cirhózy. Faktem je, že inzulinová rezistence je významně prozánětlivý metabolický stav vedoucí k produkci celé řady cytokinů, předně TNF-a, a zahajuje procesy vedoucí ke vzniku reaktivních radikálů a k peroxidaci tuků. Hormonální dysbalance charakterizovaná změnou poměru leptin/adiponektin je dalším významným faktorem vzniku převahy prozánětlivých cytokinů, která vede k expresi fenotypu Th1. Předpokládá se, že obezita je velmi významným etiologickým faktorem vzniku jaterní cirhózy, která byla dříve považována za kryptogenní.
Poměrně složitá metabolická cesta vedoucí od obezity k jaterní cirhóze samozřejmě poskytuje rozsáhlé možnosti modifikace navazujících dějů jak genetickými, tak i environmentálními a behaviorálními faktory, jejichž ovlivnění se pak stává předmětem terapeutických intervencí.
Léčba NAFLD/NASH
Přes velké rozšíření onemocnění i jeho význam existuje překvapivě málo rigorózně provedených farmakologických studií s dostatečnou statistickou silou, které by umožnily jednoznačně doporučit některý ze zkoumaných postupů. Stejně tak dietní a režimová opatření, která jsou dnes základem léčby, nejsou dostatečně podepřena empirickým důkazem a vycházejí spíše z představ o obecně správném životním způsobu a z analogie s léčbou jiných onemocnění souvisejících s obezitou a inzulinovou rezistencí [33, 34].
Dosud zkoumané léčebné postupy směřují k několika známým patofyziologickým mechanismům důležitým při vzniku a vývoji NAFLD/NASH a lze je rozdělit do dvou okruhů:
1. ‑ovlivnění inzulinové rezistence redukcí váhy, zvýšením tělesné aktivity a farmakoterapií;
2. ‑podávání hepatoprotektivních látek, především antioxidantů.
Úprava životosprávy
Základem léčby nealkoholické steatohepatitidy, tak jak je dnes doporučována, je změna životosprávy (tab. 2).

Nezbytná je pozvolná redukce váhy a tělesné cvičení. Obojí vede ke snížení obsahu tělesného tuku, zmenšení steatózy a snížení inzulinové rezistence, tedy k omezení vlivu klíčových faktorů vzniku NAFLD a následného vývoje k NASH. Redukce váhy vede jak k poklesu aktivity aminotransferáz, k úbytku obsahu tuku v jaterní tkáni, tak k ústupu zánětlivých a nekrotických změn a konečně i ke zmírnění fibrózy [35]. Žádná další léčba NASH není srovnatelně účinná s redukcí váhy a úpravou životního stylu. Nutno podotknout, že dosáhnout těchto cílů je nesnadné, adherence k restriktivnímu způsobu života je omezená a dosud jedinou léčbu obezity s prokazatelným dlouhodobým účinkem představuje bariatrická chirurgie [36]. Diety s velmi vysokým omezením energetického příjmu vhodné nejsou, jejich užívání je spojeno se zvýšením zánětlivé reakce v játrech a s nárůstem fibrózy [37]. Omezení energetického příjmu na 35–30 kcal/kg/den s cílem zredukovat váhu o 10 % v průběhu 6 měsíců se zdá být rozumné [38]. Složení diety nebylo blíže zkoumáno, většinou jsou respektována běžná doporučení omezující sacharidy a zvyšující podíl mono- a polynenasycených mastných kyselin. Problémem dietních opatření je velmi nízká a časově omezená compliance [39, 40].
Farmakoterapie
Farmakoterapie obezity u nemocných s NAFLD byla zkoumána jen okrajově. Orlistat prokázal účinnost v malé studii snížením váhy a omezením aktivity amino-transferáz [41]. Sibutramin vedl rovněž ke snížení aktivity ALT i AST v průběhu šestiměsíční léčby 13 pacientů. Problémem těchto studií je krom minimálního počtu nemocných chybění histologického průkazu vývoje onemocnění. Bariatrická chirurgie je v současnosti jedinou metodou, která významně a dlouhodoběji příznivě ovlivňuje obezitu. Její význam pro léčbu NASH je však třeba ozřejmit v rozsáhlejších studiích, protože její aplikace u morbidně obézních nemocných s jaterní lézí může vyvolat zhoršení, a to i s fatálním zakončením [42, 43].
Hypolipidemika nepřinesla obrat v léčbě NAFLD a v současnosti pouze 2 malé studie ukázaly snížení aktivit aminotransferáz při podávání atorvastatinu pacientům s NAFLD [44, 45]. Podávání fibrátů (fenofibrát, klofibrát) nepřineslo terapeutický efekt [46].
Protože centrální obezita a inzulinová rezistence hrají klíčovou úlohu při vzniku steatózy jater a při progresi NAFLD v NASH, byl nejvýznamnější terapeutický efekt očekáván od podávání látek zvyšujících citlivost tkání na inzulin. Zkoumán byl metformin a rosiglitazon. Podávání metforminu zmírnilo steatózu a snížilo aktivitu ALT v animálních studiích [38]. V otevřené randomizované studii provedené s nemocnými NASH, která srovnávala metformin 2 g/den (n = 55) s dietou (n = 27) a vita-minem E v dávkách 800 mg/den (n = 28), došlo ke statisticky významnému zlepšení ALT ve skupině pacientů užívajících metformin. Z 55 nemocných užívajících metformin byla u 17 pacientů provedena jaterní biopsie, která ukázala proti počátečnímu nálezu zmírnění steatózy, fibrózy a zánětlivých změn [47]. Thiazolidindiony (glitazony) rosiglitazon a pioglitazon se váží na PPAR-g a vedou ke zvýšení citlivosti na inzulin, ale zároveň i k redistribuci tukové tkáně. Zvyšují citlivost jaterních buněk k inzulinu, snižují aktivitu ALT i zmírňují steatózu. Byly publikovány dvě dobře navržené pilotní studie, ve kterých se podával pioglitazon v dávce 30 mg denně a rosiglitazon v dávce 4 mg 2krát denně. Došlo ke snížení aktivity ALT i k ústupu steatózy proti počátečním hodnotám, podávání pioglitazonu navíc vedlo ke zmírnění stadia fibrózy [48, 49]. Zmírnění steatózy bylo popsáno rovněž v malé randomizované studii, ve které byl pioglitazon podáván společně s vitaminem E a porovnáván s účinkem vitaminu E podávaného samostatně [50]. Velkou nevýhodou podávání thiazolidindionů je zvyšování tělesné hmotnosti ukládáním triglyceridů v periferní tukové tkáni, ke kterému dochází u 67–72 % léčených [48, 49]. Thiazolidindiony jsou navíc potenciálně hepatotoxické, což vedlo ke stažení původně zkoumaného troglitazonu z trhu [51]. V současné době je také diskutována otázka jejich nežádoucích účinků na kardiovaskulární systém [52]. Přes uvedené nedostatky jsou dnes agonisté PPAR-g pro svou schopnost zvýšit senzitivitu jaterní tkáně k inzulinu, zvyšovat oxidaci mastných kyselin a snižovat jaterní steatózu nejnadějnějšími látkami k léčbě NASH. Zkoumány byly též glitazary, které kombinují účinky na PPAR-g a PPAR-a [53]. Jejich vývoj byl v současné době ukončen.
Ve farmakoterapii NASH se uplatňují látky omezující aktivitu kyslíkových radikálů. Představa o příznivém účinku antioxidantů v léčbě NASH vychází z teorie dvojího úderu (two hit theory), kde klíčovou roli v etiopatogenezi jaterní léze a přechodu prosté steatózy v NASH hrají volné kyslíkové radikály a oxidační stres, peroxidace tuků, změny endoplazmatického retikula, lysozomální proteiny, aktivace NF-kB a aktivace prozánětlivých cytokinů. Většina publikovaných studií použila vysoké dávky vitaminu E a C s konfliktními výsledky. V současnosti chybí potřebný stupeň důkazů k všeobecnému doporučení této léčby [38]. Existuje rovněž pilotní studie s použitím pentoxifylinu jako inhibitoru produkce TNF-a, který má významnou roli v několika krocích vývoje NAFLD a NASH. U 9 z 20 léčených došlo ke zlepšení laboratorních známek jaterní léze [54].
Ursodeoxycholová kyselina nepřinesla přes počáteční slibné výsledky terapeutický efekt u pacientů s NAFLD [45]. Vzhledem k tomu, že TNF-a je aktivován bakteriálním endotoxinem, patří probiotika k potenciálně příznivě působícím látkám u nemocných s NASH. V malé studii vedlo podávání jednoho z nich (VSL 3) ke snížení aktivity ALT. Efekt na histologický nález není znám [55].
Závěr
V současnosti neexistují doporučení k léčbě NAFLD či NASH s výjimkou režimových a dietních opatření. Úloha bariatrické chirurgie u nemocných s komplikovaným NAFLD rovněž vyžaduje zevrubné zhodnocení, a není možno ji paušálně doporučit. Ostatní farmakologické léčebné postupy je možno aplikovat experimentálně, nejlépe v rámci kontrolovaných studií. Farmakoterapie přidružených komplikací syndromu inzulinové rezistence musí být prováděna s ohledem na pokročilost případné přidružené jaterní poruchy.
Seznam použité literatury
- [1] Ludwig J, Viggiano TR, McGill DB, Oh BJ. Non-alcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin Proc 1980, 55: 434–438.
- [2] Ioannou GN, Boyko EJ, Lee SP. The prevalence and predictors of elevated serum aminotransferase activity in the United States in 1999–2002. Am J Gastroenterol 2006; 101: 76–82.
- [3] McCullough AJ. The epidemiology and risk factors of NASH. In: Farrell GC, George J, de la M Hall P, McCullough AJ, eds. Fatty Liver Disease: NASH and Related Disorders. Malden, MA: Blackwell Publishing 2005: 23–37.
- [4] Farrel CF, Larter CZ. Nonalcoholic fatty liver disease: From steatosis to cirrhosis. Hepatology 2006; 43: S99–S112.
- [5] Matteoni CA, Younossi ZM, Gramlich T, et al. Nonalcoholic fatty liver disease: A spectrum of clinical and pathological severity. Gastroenterology 1999; 116: 1413–1419.
- [6] Brent A, Neuschwander-Tetri BA, Caldwell SH. Nonalcoholic steatohepatitis: summary of an AASLD single topic conference. Hepatology 2003; 37: 1202–1219.
- [7] Brunt EM, Janney CJ, Di Bisceglie AM, et al. Non-alcoholic steatohepatitis: a proposal for grading and staging the histologic lesions. Am J Gastroenterol 1999; 94: 2467–2474.
- [8] Joseph AE, Saverymuttu SH, al-Sam S, et al. Comparison of liver histology with ultrasonography in assessing diffuse parenchymal liver disease. Clin Radiol 1991; 43: 26–31.
- [9] Saadeh S, Younossi ZM, Remer EM, et al. The utility of radiological imaging in nonalcoholic fatty liver disease. Gastroenterology 2002; 123: 745–750.
- [10] Thomas EL, Hamilton G, Patel N, et al. Hepatic triglyceride content and its relation to body adiposity: a magnetic resonance imaging and proton magnetic resonance spectroscopy study. Gut 2005; 54: 122–127.
- [11] Foucher J, Chanteloup E, Vergniol J, et al. Diagnosis of cirrhosis by transient elastography (Fibroscan R): a prospective study. Gut 2005; Jul 14; PMID: 16020491. Available at: http: //gut.bmjjournals.com/cgi/rapidpdf/gut.2005.069153v1.
- [12] Rosenberg WM, Voelker M, Thiel R, et al. Serum markers detect the presence of fibrosis: a cohort study. Gastroenterology 2004; 127: 1704–1713.
- [13] Lingpunsakul S, Chalasani N. Relationship between non-alcoholic fatty liver disease and metabolit syndrome and microalbuminuria in the US adult population. Am J Med Sci 2005; 329: 111–116.
- [14] Fan JG, Zhu J, Li XJ, et al. Fatty liver and the metabolic syndrome among Shanghai adults. J Gastroenterol Hepatol 2005; 20: 1825–1832.
- [15] Hamaguchi M, Kojima T, Takeda N, et al. The metabolit syndrome as a predictor of nonalcoholic fatty liver disease. Ann Intern Med 2005; 143: 722–728.
- [16] Angulo P. Nonalcoholic fatty liver disease. N Engl J Med 2002; 16; 1221–1231.
- [17] Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest 2004; 114: 147–152.
- [18] Sanyal AJ, Campbell-Sargent C, Mirshahi F, et al. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001; 120: 1183–1192.
- [19] Samuel VT, Liu ZX, Qu X, et al. Mechanism of hepatic insulin resistence in non-alcoholic fatty liver disease. J Biol Chem 2004; 279: 32 345–32 353.
- [20] Lowell BB, Shulman GG. Mitochondrial dysfunction and type 2 diabetes. Science 2005; 307: 384–387.
- [21] Chitturi S, Abeygunasekera S, Farrell GC, et al. NASH and insulin resistance: insulin hypersecretion and specific association with the insulin resistance syndrome. Hepatology 2002; 35: 373–379.
- [22] Marchesini G, Bugianesi E, Forlani G, et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology 2003; 37: 917–923.
- [23] Feldstein AE, Canbay A, Angulo P, et al. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 2003; 125: 437–443.
- [24] Feldstein AE, Werneburg NW, Canbay A, et al. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathway. Hepatology 2004; 40: 185–194.
- [25] Pere-Carreras M, Del Hoyo P, Martin MA, et al. Defective hepatic mitochondrial respiratory Chin in patients with nonalcoholic steatohepatitis. Hepatology 2003; 38: 999–1007.
- [26] Cortez-Pinto H, Chatham J, Chatko VP, et al. Alterations in liver ATP homeostasis in human nonalcoholic steatohepatitis: a pilot study. JAMA 1999; 282: 1659–1664.
- [27] Schattenberg JM, Wang Y, Singh R, et al. Hepatocyte CYP2E1 overexpression and steatohepatitis lead to impaired hepatic insulin signaling. J Biol Chem 2005; 280: 9887–9894.
- [28] Cai D, Juan M, Frantz DF, et al. Local nad systemic resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med 2005; 11: 183–190.
- [29] Diehl AM. Tumor necrosis factor and its potential role in insulin resistence and nonalcoholic fatty liver disease. Clin Liver Dis 2004; 8: 619–638.
- [30] Saxena NK, Ikeda K, Rockey DC, et al. Leptin in hepatic fibrosis: evidence for increased collagen production in stallate cells and lean littermates of ob/ob mice. Hepatology 2002; 35: 762–771.
- [31] Ding X, Saxena NK, Lin S, et al. The role of leptin and adiponectin: a novel paradigma in adipocytokine regulation of liver fibrosis and stellate cell biology. Am J Pathol 2005; 166: 1655–1659.
- [32] Bateller R, Schwabe RF, Choi YH, et al. NADPH oxidase signal transduces angiotensin II in hepatic stellate cells and is critical in hepatic fibrosis. J Clin Invest 2003; 112: 1383–1394.
- [33] Tuomilehto J, Lindström J, Eriksson JG, et al. Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. N Engl J Med 2001; 344: 1343–1350.
- [34] Diabetes Prevention Program Research Group. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med 2002; 346: 393–403.
- [35] Dixon JB, Bhathal PS, Hughs NR, O´Brien PE. Nonalcoholic fatty liver disease: improvement in liver histological analysis with weight loss. Hepatology 2004; 39: 1647–1654.
- [36] Blackburn GL, Mun EC. Effect of weight loss surgeries. Semin Liver Dis 2004; 24: 371–379.
- [37] Andersen T, Gluud C, Franzmann MB, et al. Hepatic effects of dietary weight loss in morbidly obese subjects. J Hepatol 1991; 12: 224–229.
- [38] Adams LA, Angulo P. Treatment of non-alcoholic liver disease. Postgrad Med J 2006; 82: 315–322.
- [39] Palmer M, Schaffner F. Effect of weight reduction on hepatic abnormalities in overweight patients. Gastroenterology 1990; 99: 1408–1413.
- [40] Huang MA, Greenson JK, Chao C, et al. One-year intense nutritional counseling results in histological improvement in patients with non-alcoholic steatohepatitis: a pilot study. Am J Gastroenterol 2005; 100: 1072–1081.
- [41] Ueno T, Sugawara H, Sujaku K, et al. Therapeutic effects of restricted diet and exercise in obese patients with fatty liver. J Hepatol 1997; 27: 103–107.
- [42] Kral JG, Thung SN, Biron S, et al. Effects of surgical treatment of the metabolit syndrome on liver fibrosis and cirrhosis. Surgery 2004; 135: 48–58.
- [43] Luyckx FH, Desaive C, Thiry A, et al. Liver abnormalities in severely obese subjects: effect of drastic weight loss after gastropathy. Int J Obes Relat Metab Disord 1998; 22: 222–226.
- [44] Hatzitolios A, Savopoulos C, Lazaraki G, et al. Efficacy of omega-3 fatty acids, atorvastatin and orlistat in non-alcoholic fatty liver disease with dyslipidemia. Indian J Gastroenterol 2004; 23: 131–134.
- [45] Kiyici M, Gulten M, Gurel S, et al. Ursodeoxycholic acid and atorvastatin in the treatment of nonalcoholic steatohepatitis. Can J Gastroenterol 2003; 17: 713–718.
- [46] Laurin J, Lindor KD, Crippin JS, et al. Ursode-oxycholic acid or clofibrate in the treatment of non-alcohol-induced steatohepatitis: a pilot study. Hepatology 1996; 23: 1464–1467.
- [47] Bugianesi E, Gentilcore E, Manini R, et al. A randomized controlled trial of metformin versus vitamin E or prescriptive diet in nonalcoholic fatty liver disease. Am J Gastroenterol 2005; 100: 1082–1090.
- [48] Neuschwander-Tetri BA, Brunt EM, Wehmeier KR, et al. Improved nonalcoholic steatohepatitis after 48 weeks of treatment with the PPARgamma ligand rosiglitazone. Hepatology 2003; 38: 1008–1017.
- [49] Promrat K, Lutchman G, Uwaifo GI, et al. A pilot study of pioglitazone treatment for nonalcoholic steatohepatitis. Hepatology 2004;39:188–196.
- [50] Sanyal AJ, Mofrad PS, Contos MJ, et al. A pilot study of vitamin E versus vitamin E and pioglitazone for the treatment of nonalcoholic steatohepatitis. Clin Gastroenterol Hepatol 2004; 2: 1107–1115.
- [51] Menon KVN, Angulo P, Lindor KD. Severe cholestatic hepatitis from troglitazone in a patient with nonalcoholic steatohepatitis and diabetes mellitus. Am J Gastroenterol 2001; 96: 1631–1634.
- [52] Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med 2007; 356: 2457–2471.
- [53] Tenenbaum A, Motro M, Fisman EZ. Dual and pan-peroxisome proliferatoractivated receptors (PPAR) co-agonism: the bezafibrate lessons. Cardiovasc Diabetol 2005; 4: 14.
- [54] Adams LA, Zein CO, Angulo P, Lindor KD. A pilot trial of pentoxifylline in nonalcoholic steatohepatitis. Am J Gastroenterol 2004; 99: 2365–2368.
- [55] Loguercio C, Federico A, Tuccillo C, et al. Beneficial effects of a probiotic VSL#3 on parameters of liver dysfunction in chronic liver diseases. J Clin Gastroenterol 2005; 39: 540–543.