Nežádoucí účinky telapreviru a možnosti jejich ovlivnění
Nová éra léčby hepatitidy C s využitím trojkombinace léčiv znamená zásadní pokrok v účinnosti terapie. Na druhou stranu využití přímých antivirotik (DAA) s sebou přináší nové a častější nežádoucí účinky, které mohou interferovat s dokončením léčby a tím i s dosažením plánované účinnosti a vzácně mohou být klinicky závažné. Použití telapreviru v trojkombinační léčbě je spojeno především s vyšší incidencí rashe a pruritu, anémie a gastrointestinálních symptomů. Na základě analýzy dostupných klinických studií je však zřejmé, že aktivní přístup k časné diagnostice a řešení těchto nežádoucích účinků umožňuje jejich efektivní zvládnutí a dokončení plánované léčby a často i odeznění nežádoucích účinků ještě před ukončením podávání telapreviru (prvních 12 týdnů léčby). Znalost potenciálně závažných kožních reakcí je klíčová pro včasné ukončení léčby, což minimalizuje riziko jejich další progrese.
Standardní léčbu hepatitidy C představovala do roku 2011 kombinace pegylovaného interferonu α (PEG-IFN) a virostatika ribavirinu (RBV). Účinnost této léčby v běžné populaci dosud neléčených (tzv. naivních) pacientů není optimální a dosahuje u pacientů s genotypem 1 asi 40–45 % [1, 2]. Zásadní vliv na dosažení alespoň této efektivity má dokončení celého léčebného programu, tzn. většinou 48 týdnů této dvojkombinační léčby s optimálním dávkováním obou přípravků. Jakékoliv zkrácení léčby či snížení dávky některého z léků (většinou pod hranici 80 % předepsané dávky nebo času, tzv. pravidlo < 80 %/80 %/80 %, tedy méně než 80 % dávkování PEG-IFN a/nebo méně než 80 % dávkování RBV a/nebo méně než 80 % doporučené délky léčby) vede k signifikantnímu snížení účinnosti, obzvláště patrnému u genotypu 1, kdy se účinnost poté pohybuje kolem 35 % [3]. Léčba kombinací těchto léků je doprovázena celou řadou nežádoucích účinků (především tzv. flu-like symptomatologie, mírné kožní projevy s pruritem a ekzémy, neuropsychiatrické nežádoucí účinky a hematologické nežádoucí účinky – především významné anémie) [1, 4]. Tyto nežádoucí účinky jednak zhoršují kvalitu života pacientů a také u řady nemocných vedou buď ke snížení dávkování či k předčasnému ukončení léčby a tím snižují uvedenou účinnost. Efektivní management nežádoucích účinků kombinované léčby hepatitidy C je proto velmi důležitým procesem, který vede k optimalizaci léčby, k dosažení ideální adherence k léčbě a tím k udržení očekávané účinnosti.
Zavedení přímo působících protivirových léčiv telapreviru (TPV) a bocepreviru (BOC) – inhibitorů virových proteáz – do algoritmu léčby HCV (hepatitis C virus) infekce v roce 2011 zásadním způsobem zvýšilo účinnost terapie u pacientů s genotypem 1. Z předložených klinických studií a z registrační dokumentace obou přípravků však vyplývá, že se současně zvýšila frekvence nežádoucích účinků, některé nežádoucí účinky se prohloubily, některé se objevily poměrně nově a některé mohou být i závažné. U vyššího počtu nemocných bylo nutné léčbu předčasně ukončit. Dalšího významu nabývá tento problém u pacientů s jaterní cirhózou, což bude skupina (zvláště v našich podmínkách) zřejmě iniciálně nejčastěji léčená trojkombinační léčbou. Je tedy zásadně nutné porozumět klinickým projevům nežádoucích účinků těchto nových léčiv, včas je odhalit a znát i možnosti a způsoby jejich ovlivnění. Jedině tak můžeme optimalizovat adherenci k léčbě, umožnit dokončení léčby s optimální dávkou léku a předejít závažným zdravotním komplikacím.
V klinických studiích fáze II/III s TPV se mezi dominantními nežádoucími účinky, které se vyskytují častěji u pacientů léčených TPV (> 5% rozdíl) než u pacientů, jimž je podáváno placebo, objevily pruritus, rash, anémie a gastrointestinální potíže (anorektální dyskomfort, nauzea, průjem). Všechny tyto nežádoucí účinky lze účinně ovlivnit [5–7].
Anémie
Anémie je dobře známým nežádoucím účinkem kombinované léčby s RBV. Výskyt anémie při léčbě je považován za pozitivní prognostický faktor, který je spojen s dosažením vyšší míry setrvalé virologické odpovědi (SVR, tedy vyléčení pacienta) [8–10]. Na druhou stranu významnější anémie výrazně zhoršuje kvalitu života, eventuálně i produktivitu v zaměstnání (únava, dušnost, nevýkonnost, kognitivní problémy), může alterovat adherenci k léčbě – anémie negativně koreluje s šancí na dokončení léčby [11] a vyžaduje podle souhrnů údajů o přípravku (SPC) s obsahem RBV snížit dávkování RBV a při poklesu koncentrace hemoglobinu (Hb) pod 85 g/l ukončit podávání RBV.
Z řady studií však vyplývá, že redukce dávkování RBV při dvojkombinační léčbě může vést ke snížení SVR [3, 12]. Pokud k redukci dávkování RBV při dvojkombinační léčbě došlo již v době nedetekovatelné viremie (hodnocené jako nedetekovatelná ribonukleová kyselina viru – HCV RNA – v séru), byl negativní vliv na SVR omezený [13].
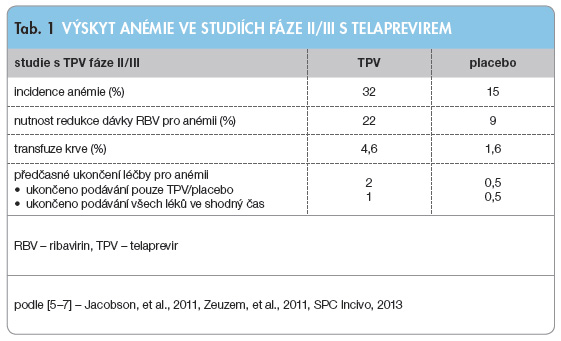
Tento problém nabývá tedy na důležitosti v případě trojkombinační léčby s TPV, kdy se anémie objevuje častěji, významněji a pokles hodnot Hb je rychlejší a hlubší (kombinace různých mechanismů vedoucích k anémii, na které se podílejí všechny tři léky), viz tab. 1. Významná anémie s koncentrací Hb < 100 g/l byla přítomna přibližně u 40 % pacientů léčených kombinací s TPV. K výraznější anémii měli predispozici především starší pacienti, pacienti s nižší v![Graf 1 Vliv anémie či redukce dávky ribavirinu na dosažení setrvalé virologické odpovědi; podle [14] – Sulkowski, et al., 2011. n/N – počet pacientů se setrvalou virologickou odpovědí/celkový počet pacientů v této skupině; PEG-IFN – pegylovaný interferon α; RBV – ribavirin; SVR – setrvalá virologická odpověď; TPV – telaprevir Léčebné režimy: PR – podávání PEG-IFN, RBV a placeba; T12PR – 12 týdnů trojkombinační léčba TPV + PEG-IFN + RBV a poté RBV + PEG-IFN](https://www.remedia.cz/photo-a-29196---.jpg) stupní hladinou Hb či pacienti s pokročilou fibrózou či cirhózou. Důležité je, že v případě trojkombinační léčby neovlivňuje významná anémie a eventuální redukce dávkování RBV v průběhu léčby dosažení SVR [14, 15], viz graf 1. SVR se neliší u pacientů s anémií a bez ní a ani snížení dávky RBV pod 600 mg/den nemělo negativní vliv na dosažení SVR ani u naivních, ani u již léčených pacientů [16]. Ve studiích s TPV nebylo dovoleno používat erytropoetin (EPO), proto byla významnější anémie korigována redukcí dávky RBV nebo podáním transfuze (u méně než 5 % pacientů). Anémii lze tedy ovlivňovat pouze manipulací s dávkou RBV, jakákoliv úprava dávky TPV či ukončení jeho podávání jsou přísně zakázány a měly by vést k ukončení celkové léčby [7]. Snížení dávky RBV v kritických prvních čtyřech týdnech trojkombinační léčby rovněž nevedlo ke snížení SVR bez ohledu na to, zda byla HCV RNA pozitivní, či negativní [16].
stupní hladinou Hb či pacienti s pokročilou fibrózou či cirhózou. Důležité je, že v případě trojkombinační léčby neovlivňuje významná anémie a eventuální redukce dávkování RBV v průběhu léčby dosažení SVR [14, 15], viz graf 1. SVR se neliší u pacientů s anémií a bez ní a ani snížení dávky RBV pod 600 mg/den nemělo negativní vliv na dosažení SVR ani u naivních, ani u již léčených pacientů [16]. Ve studiích s TPV nebylo dovoleno používat erytropoetin (EPO), proto byla významnější anémie korigována redukcí dávky RBV nebo podáním transfuze (u méně než 5 % pacientů). Anémii lze tedy ovlivňovat pouze manipulací s dávkou RBV, jakákoliv úprava dávky TPV či ukončení jeho podávání jsou přísně zakázány a měly by vést k ukončení celkové léčby [7]. Snížení dávky RBV v kritických prvních čtyřech týdnech trojkombinační léčby rovněž nevedlo ke snížení SVR bez ohledu na to, zda byla HCV RNA pozitivní, či negativní [16].
Ve studiích s BOC bylo možné použití EPO. Ani jeho časné podávání při léčbě anémie ve srovnání s redukcí dávky RBV či s oběma postupy (EPO + redukce dávky RBV) neovlivnilo celkovou SVR (byla shodná u pacientů s použitím EPO i s redukcí dávky RBV) [15]. Hlavním důvodem je zřejmě to, že stupeň dosažené anémie je odrazem vyšší expozice RBV (která je i při shodném dávkování velmi individuální), tedy anémie odráží sérovou koncentraci RBV a je markerem adekvátního dávkování, a pacientům s anémií tedy ke shodné účinnosti postačuje nižší dávka RBV. Pacienty, jimž je podávána trojkombinační léčba, je třeba sledovat častěji, zvláště krevní obraz je vhodné kontrolovat alespoň každé dva týdny. V případě rychlého či hlubokého poklesu hladiny Hb (v závislosti na eventuální přítomnosti srdečního onemocnění), či pokud dojde k poklesu koncentrace Hb na méně než 100 g/l, je třeba snížit dávku RBV na 600 mg/den a po stabilizaci koncentrace Hb se pokusit pomalu dávku zvyšovat. Tento agresivní přístup s rychlým poklesem dávkování RBV se zdá výhodnější než jeho pozvolná úprava. V případě poklesu hladiny Hb pod 85 g/l je vhodné podávání RBV dočasně ukončit a po vzestupu koncentrace Hb je znovu zahájit v nižší dávce, kterou lze opět titrovat dle laboratorní odpovědi.
Doporučení pro management anémie při trojkombinační léčbě s TPV
Léčebný postup ke zvládnutí anémie při trojkombinační léčbě s TPV, který by neměl ovlivnit dosažení SVR [7, 14, 17]:
- monitorovat krevní obraz v týdnu 2, 4, 6 a dále minimálně každé 4 týdny (během trojkombinační léčby lépe každé 2 týdny);
- při poklesu koncentrace Hb pod 100 g/l snížit dávku RBV na 600 mg;
- při vzestupu hladiny Hb titrovat dávku RBV – většinou do dávky 800 mg/den, eventuálně je při dalším poklesu koncentrace Hb možné titrovat opačně – dávku snižovat – pod hodnotu 600 mg/den;
- při poklesu koncentrace Hb pod 85 g/l je doporučeno podávání RBV dočasně ukončit;
- pokud je RBV trvale (déle než 14 dní) vysazen, podávání TPV musí být rovněž ukončeno a nesmí být znovu zahájeno [7];
- po vzestupu hladiny Hb znovu započít s podáváním RBV a dle laboratorní odpovědi dávku titrovat;
- podávání EPO či krevní transfuze rezervovat pouze pro nemocné, u kterých selže tento postup.
Dermatologické nežádoucí účinky
Kožní nežádoucí reakce při léčbě PEG-IFN/RBV jsou známé a dobře popsané a jsou zodpovědné za více než 10 % všech nežádoucích účinků asociovaných s podáváním IFN [18]. Většinou jde o kožní projevy, které lze zařadit do skupiny dermatitid. Jedná se především o generalizovaný pruritus a suchost kůže, dále jsou přítomny léze podobné ekzému, které mohou progredovat až do začervenalých papul a mikropuchýřků s možností exkoriace (oděrek) [19]. Tyto projevy lze léčit podobně jako jiné ekzémy, tzn. lokálně (topicky) aplikovat přípravky s obsahem kortikosteroidů, které jsou postupně nahrazeny emoliencii (látky zvyšující hydrataci kůže doplněním vody do rohové vrstvy kůže, např. bílá vazelína, parafínový olej, stearylalkohol). Je možné použít i systémová antihistaminika. Tyto potíže jsou většinou léčbou ovlivnitelné a nevyžadují přerušení či ukončení protivirové léčby.
Nová éra v léčbě hepatitidy C s využitím přímých antivirotik (directly acting antivirals, DAA) však staví ošetřující lékaře před změnu klinického charakteru kožních nežádoucích účinků, zvýšení frekvence i zvýšení jejich významnosti včetně potenciálně život ohrožujících kožních komplikací. Současně jsou tyto nežádoucí účinky natolik významné, že mohou vést k úpravě dávkování léčby a těžký průběh kožních nežádoucích účinků znamená nutnost ukončení kombinované léčby. Znalost klinické manifestace a možností léčby kožních nežádoucích účinků je proto zásadní pro optimální průběh terapie HCV infekce.
V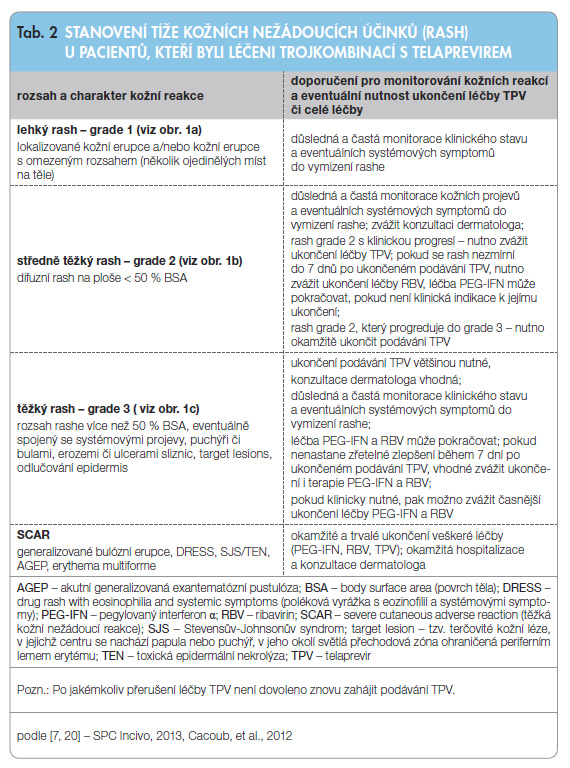 registračních klinických studiích byla frekvence kožních nežádoucích účinků ve studiích s TPV vyšší ve srovnání s placebem [5–7]. Ve studiích fáze II/III s TPV dostalo celkem 2012 pacientů minimálně jednu dávku TPV a 764 pacientů minimálně jednu dávku placeba. U celkem 55 % pacientů užívajících TPV se objevil rash, zatímco u pacientů s placebem v 33 % případů [7]. Rash byl u pacientů užívajících TPV těžší a rozsáhlejší, ale svojí charakteristikou byl velmi podobný vyrážce při léčbě PEG-IFN/RBV (tedy bez TPV) jak vizuálně, tak histopatologicky. Rash byl především svědivý, někdy ekzematózního charakteru či s makulopapulární složkou. Histologicky se jedná
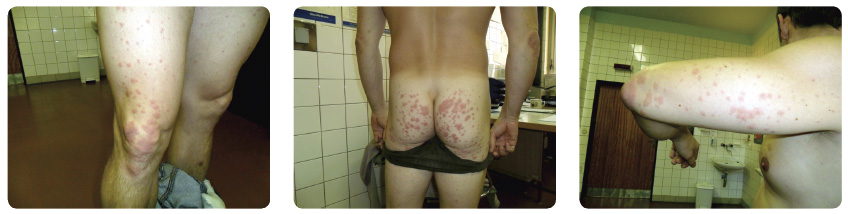
registračních klinických studiích byla frekvence kožních nežádoucích účinků ve studiích s TPV vyšší ve srovnání s placebem [5–7]. Ve studiích fáze II/III s TPV dostalo celkem 2012 pacientů minimálně jednu dávku TPV a 764 pacientů minimálně jednu dávku placeba. U celkem 55 % pacientů užívajících TPV se objevil rash, zatímco u pacientů s placebem v 33 % případů [7]. Rash byl u pacientů užívajících TPV těžší a rozsáhlejší, ale svojí charakteristikou byl velmi podobný vyrážce při léčbě PEG-IFN/RBV (tedy bez TPV) jak vizuálně, tak histopatologicky. Rash byl především svědivý, někdy ekzematózního charakteru či s makulopapulární složkou. Histologicky se jedná 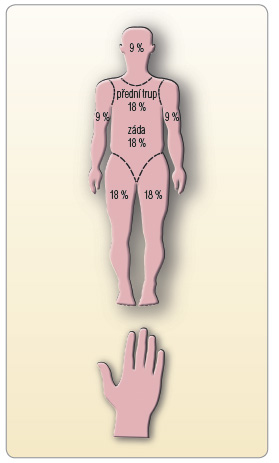 především o spongiformní dermatitidu s dominantně lymfatickou či eozinofilní perivaskulární infiltrací. Většina případů rashe byla mírná či středně těžká (grade 1 či 2) a progrese do těžkého rashe (grade 3) byla ojedinělá (< 10 % případů) [7, 20]. Znalost klasifikace tíže rashe je klinicky velmi důležitá pro stanovení dalšího léčebného postupu. Rozdělení rashe dle jeho tíže a doporučení týkající se dalších opatření či léčby jsou uvedeny v tab. 2 a typická kožní manifestace je patrná na obr. 1a–c
především o spongiformní dermatitidu s dominantně lymfatickou či eozinofilní perivaskulární infiltrací. Většina případů rashe byla mírná či středně těžká (grade 1 či 2) a progrese do těžkého rashe (grade 3) byla ojedinělá (< 10 % případů) [7, 20]. Znalost klasifikace tíže rashe je klinicky velmi důležitá pro stanovení dalšího léčebného postupu. Rozdělení rashe dle jeho tíže a doporučení týkající se dalších opatření či léčby jsou uvedeny v tab. 2 a typická kožní manifestace je patrná na obr. 1a–c![Obr. 1a, b, c Typické projevy rashe u pacientů léčených telaprevirem: a – lehký, b – středně těžký, c – těžký; podle [20, 28] – Cacoub, et al., 2012, Janssen, 2012.](https://www.remedia.cz/photo-a-29199---.jpg) . K orientačnímu odhadu rozsahu postižení kůže je vhodné znát procentuální rozložení kožního povrchu na jednotlivých částech těla, viz obr. 2.
. K orientačnímu odhadu rozsahu postižení kůže je vhodné znát procentuální rozložení kožního povrchu na jednotlivých částech těla, viz obr. 2.
Rash se v polovině případů objevil již v prvních 4 týdnech kombinované léčby. Zbylé případy rashe (50 %) se postupně objevovaly mezi 5. a 12. týdnem léčby. Těžký rash (grade 3, s nutností zvážení ukončení léčby TPV), který je ekzematózní, svědivý, s možnou makulopapulární složkou, s možnými systémo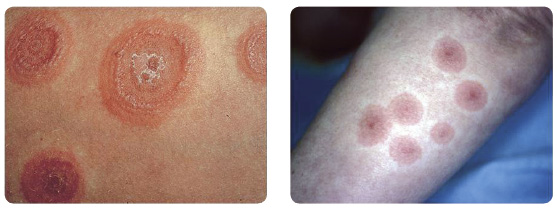 vými projevy či erozemi a ulcerami sliznic a zahrnující více než 50 % plochy těla, se vyskytl celkem u 4,8 % pacientů léčených TPV a u 0,4 % pacientů léčených PEG-IFN/RBV a placebem [7]. Důležité je srovnání výskytu rashe a nutnosti předčasného ukončení léčby v důsledku kožních nežádoucích účinků u pacientů ve studiích fáze II (PROVE 1–3) a ve studiích fáze III (ADVANCE a REALIZE). Ve studiích fáze II došlo k předčasnému ukončení léčby v důsledku výskytu rashe u 6 % pacientů léčených TPV (35 pacientů z 566) oproti 0,4 % pacientů (1 pacient z 277) léčených placebem (dvojkombinační léčba). Naproti tomu ve studiích fáze III došlo k předčasnému ukončení léčby v důsledku rashe u 1 % nemocných léčených TPV (13 pacientů z 1257) a u žádného pacienta, kterému bylo podáváno placebo (resp. dvojkombinační léčba) [21].
vými projevy či erozemi a ulcerami sliznic a zahrnující více než 50 % plochy těla, se vyskytl celkem u 4,8 % pacientů léčených TPV a u 0,4 % pacientů léčených PEG-IFN/RBV a placebem [7]. Důležité je srovnání výskytu rashe a nutnosti předčasného ukončení léčby v důsledku kožních nežádoucích účinků u pacientů ve studiích fáze II (PROVE 1–3) a ve studiích fáze III (ADVANCE a REALIZE). Ve studiích fáze II došlo k předčasnému ukončení léčby v důsledku výskytu rashe u 6 % pacientů léčených TPV (35 pacientů z 566) oproti 0,4 % pacientů (1 pacient z 277) léčených placebem (dvojkombinační léčba). Naproti tomu ve studiích fáze III došlo k předčasnému ukončení léčby v důsledku rashe u 1 % nemocných léčených TPV (13 pacientů z 1257) a u žádného pacienta, kterému bylo podáváno placebo (resp. dvojkombinační léčba) [21].
 Lze tedy zaznamenat poměrně zásadní pokles nutnosti přerušení léčby u pacientů léčených TPV z důvodu kožních nežádoucích účinků mezi studiemi fáze II a fáze III. Tohoto cíle bylo zřejmě dosaženo v důsledku nastavení přesného plánu diagnostiky a časné léčby rashe, který byl implementován v protokolech všech studií fáze III [5, 6]. Tento tzv. rash management plan poskytuje jasné doporučení, jak postupovat v případě výskytu pruritu a rashe. Je prokázáno, že aktivní léčebný přístup v časných stadiích rashe vede ke stabilizaci této dermatologické komplikace, zabraňuje progresi do těžších stadií (včetně klinicky závažných), vede ke stabilizaci nálezu a většinou i k regresi rashe ještě před koncem léčby TPV (viz obr. 4, 5, 6). Plán diagnostiky i zvládnutí rashe (viz tab. 2) [5, 6, 20] poskytuje jasný návod pro lékaře i pacienty, jak postupovat při zvládání nežádoucích kožních reakcí při léčbě s TPV, a tím umožňuje optimalizovat terapii a dosáhnout ideálního výsledku léčby. Vždy je nutné mít na mysli, že v případě přerušení či ukončení léčby s TPV není nové podávání léku dovoleno a je možné pokračovat pouze v dvojkombinační léčbě PEG-IFN + RBV [7] (pokud není nutné z klinických důvodů ukončit i tuto léčbu).
Lze tedy zaznamenat poměrně zásadní pokles nutnosti přerušení léčby u pacientů léčených TPV z důvodu kožních nežádoucích účinků mezi studiemi fáze II a fáze III. Tohoto cíle bylo zřejmě dosaženo v důsledku nastavení přesného plánu diagnostiky a časné léčby rashe, který byl implementován v protokolech všech studií fáze III [5, 6]. Tento tzv. rash management plan poskytuje jasné doporučení, jak postupovat v případě výskytu pruritu a rashe. Je prokázáno, že aktivní léčebný přístup v časných stadiích rashe vede ke stabilizaci této dermatologické komplikace, zabraňuje progresi do těžších stadií (včetně klinicky závažných), vede ke stabilizaci nálezu a většinou i k regresi rashe ještě před koncem léčby TPV (viz obr. 4, 5, 6). Plán diagnostiky i zvládnutí rashe (viz tab. 2) [5, 6, 20] poskytuje jasný návod pro lékaře i pacienty, jak postupovat při zvládání nežádoucích kožních reakcí při léčbě s TPV, a tím umožňuje optimalizovat terapii a dosáhnout ideálního výsledku léčby. Vždy je nutné mít na mysli, že v případě přerušení či ukončení léčby s TPV není nové podávání léku dovoleno a je možné pokračovat pouze v dvojkombinační léčbě PEG-IFN + RBV [7] (pokud není nutné z klinických důvodů ukončit i tuto léčbu).
 Vždy tedy musíme dopředu počítat s rizikem výskytu těžšího pruritu či rashe při trojkombinační léčbě s TPV. Je nutné věnovat zásadní pozornost prevenci kožních nežádoucích účinků, tzn. pečlivé péči o kůži, její hydrataci a zabránění vlivu jiných podnětů dráždících kůži. Léčbu rashe můžeme rozdělit na prevenci, léčbu lokální a léčbu celkovou. Doporučené postupy a přípravky pro symptomatickou terapii jednotlivých fází kožních reakcí jsou uvedeny v tab. 3. Systémové užití kortikosteroidů není vhodné, neboť může snižovat účinnost kombinované protivirové léčby.
Vždy tedy musíme dopředu počítat s rizikem výskytu těžšího pruritu či rashe při trojkombinační léčbě s TPV. Je nutné věnovat zásadní pozornost prevenci kožních nežádoucích účinků, tzn. pečlivé péči o kůži, její hydrataci a zabránění vlivu jiných podnětů dráždících kůži. Léčbu rashe můžeme rozdělit na prevenci, léčbu lokální a léčbu celkovou. Doporučené postupy a přípravky pro symptomatickou terapii jednotlivých fází kožních reakcí jsou uvedeny v tab. 3. Systémové užití kortikosteroidů není vhodné, neboť může snižovat účinnost kombinované protivirové léčby.
Z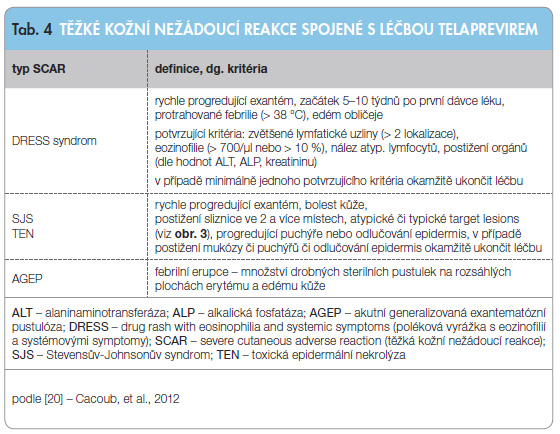 klinického hlediska je nezbytné znát definici těžkých nežádoucích kožních reakcí (severe cutaneous adverse reaction, SCAR), poněvadž rozvoj a progrese těchto kožních změn může mít závažný průběh včetně možného fatálního vyústění (pokud jsou pozdě rozpoznány či neadekvátně léčeny). Přehled jednotlivých typů SCAR je uveden v tab. 4 [20]. Tyto těžké kožní reakce se v klinických studiích nevyskytly příliš často, jejich průběh byl ale závažný. Celkově se podezření na DRESS (Drug Rash with Eosinophilia and Systemic Symptoms) syndrom objevila v 11 případech (0,4 %), podezření na Stevensův-Johnsonův syndrom (SJS) ve 3 případech (0,1 %). Obecná mortalita u těchto kožních komplikací je vysoká (13 % při SJS až 39 % při toxické epidermální nekrolýze, TEN) [22, 23]. DRESS syndrom je méně agresivní komplikací s mortalitou kolem 10 % [24]. V klinických studiích s TPV nebylo ale zaznamenáno žádné úmrtí na tyto kožní komplikace. V současné době je pravděpodobné, že se v klinické praxi několik fatálních průběhů (hlášených farmakovigilančním autoritám) objevilo. Dle hlášení se jednalo o pacienty s progredujícím rashem a systémovými projevy (teploty, nauzea, průjem, slizniční ulcerace, otok obličeje, konjunktivitida), kteří pokračovali v trojkombinační léčbě v nezměněném dávkování (neboli léčba s TPV nebyla včas ukončena) [25].
klinického hlediska je nezbytné znát definici těžkých nežádoucích kožních reakcí (severe cutaneous adverse reaction, SCAR), poněvadž rozvoj a progrese těchto kožních změn může mít závažný průběh včetně možného fatálního vyústění (pokud jsou pozdě rozpoznány či neadekvátně léčeny). Přehled jednotlivých typů SCAR je uveden v tab. 4 [20]. Tyto těžké kožní reakce se v klinických studiích nevyskytly příliš často, jejich průběh byl ale závažný. Celkově se podezření na DRESS (Drug Rash with Eosinophilia and Systemic Symptoms) syndrom objevila v 11 případech (0,4 %), podezření na Stevensův-Johnsonův syndrom (SJS) ve 3 případech (0,1 %). Obecná mortalita u těchto kožních komplikací je vysoká (13 % při SJS až 39 % při toxické epidermální nekrolýze, TEN) [22, 23]. DRESS syndrom je méně agresivní komplikací s mortalitou kolem 10 % [24]. V klinických studiích s TPV nebylo ale zaznamenáno žádné úmrtí na tyto kožní komplikace. V současné době je pravděpodobné, že se v klinické praxi několik fatálních průběhů (hlášených farmakovigilančním autoritám) objevilo. Dle hlášení se jednalo o pacienty s progredujícím rashem a systémovými projevy (teploty, nauzea, průjem, slizniční ulcerace, otok obličeje, konjunktivitida), kteří pokračovali v trojkombinační léčbě v nezměněném dávkování (neboli léčba s TPV nebyla včas ukončena) [25].
Je nezbytně nutné při jakémkoliv podezření na závažnou polékovou kožní reakci (SCAR) okamžitě ukončit veškerou terapii, pacienti musí být neprodleně hospitalizováni a musí být konzultován dermatolog [20]. O symptomatologii kožních projevů a nutnosti okamžitého hlášení kožních nežádoucích účinků ošetřujícímu lékaři musí být pacienti před zahájením léčby prokazatelně poučeni.
Anorektální symptomatologie
V kontrolovaných registračních studiích fáze II/III se anorektální problémy (svědění, rektální bolest, pálení v konečníku, hemoroidy) vyskytovaly významně častěji u pacientů léčených TPV (26,2 % vs. 5,4 %) [5–7]. Potíže většinou začínaly v prvních dvou týdnech léčby. Většinou se jednalo o potíže mírné či středně těžké, které v podstatě nevedly k přerušení či ukončení léčby a většinou odezněly po ukončení podávání TPV. Mechanismus této komplikace zůstává neobjasněný a také není zjevná spojitost s výskytem pruritu či kožního rashe. V rámci léčby by mělo být provedeno vyšetření anální oblasti k vyloučení jiných ovlivnitelných lézí, které by mohly vysvětlovat symptomy (hemoroidy, fisury či píštěle). V případě absence jiných ovlivnitelných příčin je indikována běžná symptomatická léčba s použitím lokálních přípravků (včetně lokálních anestetik, např. 3% lidokainu, kortikosteroidů) či systémových antihistaminik.
Bezpečnost kombinované léčby s TPV u pacientů s jaterní cirhózou
Přítomnost jaterní cirhózy představuje negativní prediktivní faktor úspěšnosti dvojkombinační léčby. Trojkombinační léčba s TPV výrazně zvýšila šance pacientů s jaterní cirhózou na dosažení SVR (tedy vyléčení). Pacienti s HCV jaterní cirhózou (a kteří neodpověděli na již podanou dvojkombinační léčbu) budou zvláště v našich podmínkách patřit mezi ty nemocné, kterým bude trojkombinační léčba podávána nejdříve (zdravotní indikace, domluva se zdravotními pojištovnami). V registračních studiích fáze III byli pacienti vybíráni podle přesně definovaných vstupních a vylučovacích kritérií, přičemž klinické či laboratorní známky pokročilejšího jaterního onemocnění patřily mezi vylučovací kritéria. Nežádoucí účinky (rash, pruritus, anémie) se v těchto registračních studiích vyskytovaly častěji u nemocných s cirhózou a léčených TPV než u pacientů bez cirhózy léčených TPV (rash 67 % vs. 52 %, pruritus u 53 % vs. 42 % a anémie u 59 % vs. 34 %). Nežádoucí účinky vedly k předčasnému ukončení léčby u 15 % pacientů s TPV a cirhózou oproti 11 % pacientů s TPV bez cirhózy [26].
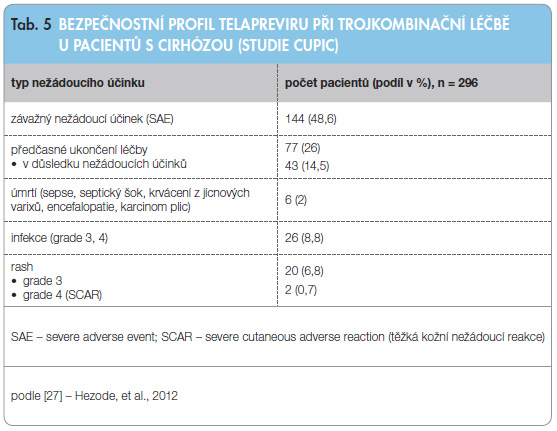 Z důvodu ověření bezpečnosti podávání DAA v trojkombinační léčbě u pacientů s cirhózou v reálné klinické praxi byly ve Francii shromážděny údaje v tzv. CUPIC kohortě. Cílem bylo určit účinnost a bezpečnost léčby, rezistenci vůči DAA, farmakokinetiku DAA a kvalitu života pacientů [27]. Celkem bylo do této studie zařazeno 674 pacientů s cirhózou a genotypem 1 léčených trojkombinační léčbou, k analýze bylo vybráno 455 pacientů, kteří byli léčeni minimálně 16 týdnů. Z této skupiny bylo 296 pacientů léčeno trojkombinací s TPV (v běžném dávkování). Většina pacientů se nacházela ve stadiu Child A, ale 15 % ze souboru již mělo jícnové varixy a celkem 34 % pacientů léčených TPV splňovalo vylučovací kritéria studií fáze III. Tato populace nemocných tedy lépe odráží skutečnou skupinu pacientů s pokročilým jaterním onemocněním, kteří budou léčeni trojkombinační léčbou. Shrnutí informací týkajících se nežádoucích účinků u těchto nemocných je uvedeno v tab. 5.
Z důvodu ověření bezpečnosti podávání DAA v trojkombinační léčbě u pacientů s cirhózou v reálné klinické praxi byly ve Francii shromážděny údaje v tzv. CUPIC kohortě. Cílem bylo určit účinnost a bezpečnost léčby, rezistenci vůči DAA, farmakokinetiku DAA a kvalitu života pacientů [27]. Celkem bylo do této studie zařazeno 674 pacientů s cirhózou a genotypem 1 léčených trojkombinační léčbou, k analýze bylo vybráno 455 pacientů, kteří byli léčeni minimálně 16 týdnů. Z této skupiny bylo 296 pacientů léčeno trojkombinací s TPV (v běžném dávkování). Většina pacientů se nacházela ve stadiu Child A, ale 15 % ze souboru již mělo jícnové varixy a celkem 34 % pacientů léčených TPV splňovalo vylučovací kritéria studií fáze III. Tato populace nemocných tedy lépe odráží skutečnou skupinu pacientů s pokročilým jaterním onemocněním, kteří budou léčeni trojkombinační léčbou. Shrnutí informací týkajících se nežádoucích účinků u těchto nemocných je uvedeno v tab. 5.
Z tabulky je patrné, že incidence závažných nežádoucích účinků (48,6 %) byla vyšší než ve studiích fáze III. Také předčasné ukončení léčby (v tomto případě do 16. týdne léčby) bylo častější (26 %), z důvodu nežádoucích účinků k němu došlo u 14,5 % nemocných. Během 16 týdnů bylo zaznamenáno 6 úmrtí (2 %), z toho polovina následkem těžké infekce a sepse. Těžká infekce se vyskytla celkem u 8,8 % pacientů. K dekompenzaci jaterní cirhózy došlo u 4,4 % pacientů. Kromě těchto klinických komplikací byly časté i hematologické nežádoucí účinky. K poklesu koncentrace Hb pod 100 g/l došlo u 29,7 % pacientů a k poklesu pod 80 g/l u 10,1 % pacientů. Trombocytopenie s poklesem počtu krevních destiček pod 50 000/mm3 se objevila u 11,8 % pacientů. Analýza výsledků pacientů, kteří během této studie zemřeli či vyvinuli těžkou infekci, ukazuje, že rizikem rozvoje závažné (eventuálně život ohrožující) infekce jsou ohroženi především nemocní s anamnézou jaterní dekompenzace, s klasifikací Child-Pugh score > 5, s portální hypertenzí, s nízkou vstupní hladinou trombocytů. Tito pacienti jednoznačně vyžadují velice časté monitorování laboratorních hodnot a klinického stavu a časnou intervenci.
Podpořeno projektem Ministerstva zdravotnictví koncepčního rozvoje výzkumné organizace 00669806 – FN Plzeň.
Seznam použité literatury
- [1] Fried MW, Shiffman ML, Reddy KR, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med 2002; 347: 975–982.
- [2] Manns MP, McHutchison JG, Gordon SC, et al. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet 2001; 358: 958–965.
- [3] McHutchison JG, Manns M, Patel K, et al. Adherence to combination therapy enhances sustained response in genotype-1-infected patients with chronic hepatitis C. Gastroenterology 2002; 123: 1061–1069.
- [4] EASL Clinical Practice Guidelines: management of hepatitis C virus infection. J Hepatol 2011; 55: 245–264.
- [5] Jacobson IM, McHutchison JG, Dusheiko G, et al. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 2011; 364: 2405–2416.
- [6] Zeuzem S, Andreone P, Pol S, et al. Telaprevir for retreatment of HCV infection. N Engl J Med 2011; 364: 2417–2428.
- [7] European Medicines Agency. Telaprevir (Incivo): EU summary of product characteristics [online]. Available from URL: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002313/WC500115529.pdf [accesed 2013 Feb 25].
- [8] Sulkowski MS, Shiffman ML, Afdhal NH, et al. Hepatitis C virus treatment-related anemia is associated with higher sustained virologic response rate. Gastroenterology 2010; 139: 1602–1611.
- [9] Sievert W, Dore GJ, McCaughan GW, et al. Virological response is associated with decline in hemoglobin concentration during pegylated interferon and ribavirin therapy in hepatitis C virus genotype 1. Hepatology 2011; 53: 1109–1117.
- [10] McHutchison JG, Lawitz EJ, Shiffman ML, et al. Peginterferon alfa-2b or alfa-2a with ribavirin for treatment of hepatitis C infection. N Engl J Med 2009; 361: 580–593.
- [11] Butt AA, McGinnis KA, Skanderson M, et al. Hepatitis C treatment completion rates in routine clinical care. Liver Int 2010; 30: 240–250.
- [12] Lo RV III, Teal V, Localio AR, et al. Relationship between adherence to hepatitis C virus therapy and virologic outcomes: a cohort study. Ann Intern Med 2011; 155: 353–360.
- [13] Reddy KR, Shiffman ML, Morgan TR, et al. Impact of ribavirin dose reductions in hepatitis C virus genotype 1 patients completing peginterferon alfa-2a/ribavirin treatment. Clin Gastroenterol Hepatol 2007; 5: 124–129.
- [14] Sulkowski MS, Reddy R, Afdhal NH, et al. Anemia had no effect on efficacy outcomes in treatment-naive patients who received telaprevir-based regimen in the advance and illuminate phase 3 studies. J Hepatol 2011; 54 (suppl.1), S195.
- [15] Poordad FF, Lawitz EJ, Reddy KR, et al. A randomized trial comparing ribavirin dose reduction versus erythropoietin for anaemia management in previously untreated patients with chronic hepatitis C receiving boceprevir plus peginterferon/ribavirin (Abstract). J Hepatol 2012; 56 (Suppl.2): S559.
- [16] Sulkowski MS, Roberts S, Afdhal N, et al. Ribavirin dose modification in treatment-naive and previously treated patients who received telaprevir combination treatment: no impact on sustained virologic response in phase III studies. J Hepatol 2012; 56 (Suppl. 1): S459– S460.
- [17] Roberts S, Andreone P, Pol S, et al. Impact of Anemia and Ribavirin Dose Reduction on SVR to a Telaprevir-based Regimen in Patients with HCV Genotype 1 and Prior Peginterferon/Ribavirin Treatment Failure in the Phase III REALIZE Study. Presented at the Annual Meeting of the American Associtation for the Study of Liver Diseases 2011 November 4-8, 2011 San Francisco, CA, Abstract 1366.
- [18] Charron A, Bessis D, Dereure O, et al. Local cutaneous side effects of interferons. Presse Med 2001; 30: 1555–1560.
- [19] Lubbe J, Kerl K, Negro F, et al. Clinical and immunological features of hepatitis C treatment-associated dermatitis in 36 prospective cases. Br J Dermatol 2005; 153: 1088–1090.
- [20] Cacoub P, Bourliere M, Lubbe J, et al. Dermatological side effects of hepatitis C and its treatment: patient management in the era of direct-acting antivirals. J Hepatol 2012; 56: 455–463.
- [21] FDA Antiviral Drugs Advisory Committee. http://www.fda.gov/downloads/Advisory-Committees/Committees/MeetingMaterials/Drugs/AntiviralDrugsAdvisory-Committee/UCM252562.pdf. 28-4-2011. [Accesed 2013 Feb 25].
- [22] Roujeau JC, Stern RS. Severe adverse cutaneous reactions to drugs. N Engl J Med 1994; 331: 1272–1285.
- [23] Roujeau JC, Allanore L, Liss Y, et al. Severe Cutaneous Adverse Reactions to drugs (SCAR): definitions, diagnostic criteria, genetic predisposition. Dermatol Sinica 2009; 27: 203–209.
- [24] Cacoub P, Musette P, Descamps V, et al. The DRESS syndrome: a literature review. Am J Med 2011; 124: 588–597.
- [25] FDA Drug Safety Comunication. http://www.fda.gov/Drugs/DrugSafety/ucm332731.htm. 2012 Dec 19. [Accessed 2013 Feb 25].
- [26] Pol S, Roberts SK, Andreone P. Efficacy and safety of telaprevir-based regimen in cirrhotic patients with HCV genotype-1 and prior peginterferon/ribavirin treatment failure: subanalysis of the REALIZE phase III study. Hepatology 2011; 54 (Suppl. S1): 374A.
- [27] Hezode C, Dorival C, Zoulim F, et al. Safety of telaprevir or boceprevir in combination with peginterferon alfa/ribavirin in cirrhotic non responders. First results of the French early access program (ANRS CO20-CUPIC). J Hepatol 2012; 56 (Suppl.2): S4.
- [28] Postup při řešení dermatologických nežádoucích účinků pacientů léčených přípravkem INCIVO. Informační brožura společnosti Janssen, 2012.