Originální a generické léčivé přípravky, biologická a biologickým lékům podobná léčiva (biosimilars) a nebiologická komplexní léčiva
Souhrn:
Vedle generických přípravků a tzv. biosimilars se objevila nová skupina léčiv, tzv. nebiologická komplexní léčiva, která mají zcela odlišné charakteristiky a východiska pro registraci a další regulace, jako je úhrada z veřejných prostředků. Správné pochopení rozdílů mezi těmito léky, které jsou podobné originál-ním přípravkům, je u lékařů důležité pro náležitou léčbu pacientů, u regulačních orgánů pak pro volbu správných postupů a algoritmů; jejich výsledkem je možnost uvádět taková léčiva na trh.
Key words: original drugs – generics – biosimilars – non biological complex drugs – glatiramer – glatiramoids.
Summary:
Beside generic drugs and so called biosimilars, a new drug class was developed – so called non biological complex drugs. Non biological complex drugs have completely different characteristics and basis for registration and other regulations, including the coverage from public financial sources. Proper understanding of the differences among the above drug classes – similar to original drugs – is crucial for the physicians so that they are able to adequately treat their patients and for the regulatory organs, too, so that they can choose correct procedures and algorithms. When all these conditions are met, the new drug classes can be marketed.
Úvod
V současné době jsou v zemích Evropské unie (EU) vedle originálních léčivých přípravků v široké míře dostupná tzv. generika (generické lékové ekvivalenty) nebo tzv. podobná biologická léčiva (biosimilars).
Generika jsou přípravky obsahující stejnou účinnou látku jako originální přípravek, mohou vstoupit na trh po vypršení patentové ochrany originálních léčivých přípravků. Obvykle vycházejí z léčiv s malou molekulou vyráběnou povětšinou synteticky. Biosimilars naproti tomu vycházejí z originálních léčiv se středně velkou až velkou molekulou vyráběnou obvykle rekombinantní technologií (vložením příslušného genomu do vhodných buněk), kde biologická povaha vylučuje stoprocentní shodu ve struktuře komplexní molekuly.
Zatímco generické přípravky v řadě zemí již předstihly v počtu balení originální léčivé přípravky, v případě biosimilars jsme na začátku jejich cesty, i když jsou některá z nich dostupná řadu let. Vedle toho se blíží vstup nové skupiny léčiv, tzv. nebiologických komplexních léčiv („glatiramoidů“, tedy látek podobných glatirameru), která nesplňují znaky zákonné definice generika nebo biosimilars. Autor si dovoluje uvést své názory na problémy spojené s případným vstupem skupiny glatiramoidů na trh v České republice (ČR).
Originální a generické přípravky byly v minulosti opakovaně předmětem podrobného popisu v tuzemských odborných periodikách [1,2], a i když autoři nesdíleli vždy zcela totožné názory, pokaždé se shodli na užitečnosti souběžné existence obou skupin. Ačkoliv titulky webových vyhledávačů mohou naznačovat, že mezi originály a generiky probíhá lítý boj, ve skutečnosti velká část farmaceutických společností, zejména těch větších, vyrábí a na trh uvádí obě tyto skupiny přípravků.
Existence generik by se v budoucnosti mohla stát dalším nebo novým impulsem pro změnu role lékárníků a lékařů [3], ovšem za předpokladu, že dojde ke zlepšení kvality informací a spolupráce všech zainteresovaných subjektů, zejména zdravotních pojišťoven. Implementace generických přípravků nepochybně vede ke snížení výdajů z veřejných prostředků a k případnému zvýšení počtu léčených osob, jak bylo nedávno velmi pěkně doloženo na příkladu statinů pro ČR [4]. Na druhou stranu je velmi obtížné definitivně zhodnotit klinickou účinnost generik v podmínkách běžné klinické praxe. Z publikovaných prací totiž plyne jak účinnost vyšší [5], tak i nižší [6], případně nemusejí být zjištěny rozdíly žádné [7], a při troše „dobré vůle“ tak lze argumentaci vždy posunout k „žádoucímu cíli“.
Od roku 2012 dokonce vychází nové odborné periodikum s názvem Generics and Biosimilars Initiative Journal [8], jehož cílem je publikování nezávislých vědeckých pojednání o generických přípravcích a biosimilars z hlediska jejich vývoje, výroby, testování (včetně bioekvivalence), regulace, farmakoekonomiky a distribuce. Kromě toho časopis usiluje o publikování zkrácených verzí příslušných regulačních směrnic a právních předpisů tak, aby se tyto dokumenty mohly dostat ke všem zájemcům.
Právní úprava
Generika
Zatímco platná právní úprava definuje generikum (ustanovení § 25 odst. 4 písm. b) zákona č. 378/2007 Sb., o léčivech) jako „léčivý přípravek, který má shodné kvalitativní a kvantitativní složení, pokud jde o léčivé látky, a shodnou lékovou formu s referenčním léčivým přípravkem a u kterého byla, s výjimkou případů, kdy lze doložit, že generikum splňuje příslušná kritéria stanovená příslušnými pokyny Komise a agentury, prokázána bioekvivalence s referenčním léčivým přípravkem příslušnými studiemi biologické dostupnosti; různé soli, estery, ethery, izomery, směsi izomerů, komplexy nebo deriváty léčivé látky se považují za tutéž léčivou látku, pokud se významně neodlišují vlastnostmi týkajícími se bezpečnosti, popřípadě účinnosti; různé perorální lékové formy s okamžitým uvolňováním se považují za jednu a tutéž lékovou formu.“, zákonná definice originálního přípravku neexistuje. Dokonce je možné registrovat generikum dle referenčního přípravku, který je též generikem, přičemž příslušný referenční přípravek dokonce nemusí být v ČR ani registrován.
Biosimilars
Podobně v případě tzv. podobných biologických léčiv (autor raději užívá výraz biosimilars) není definován originální přípravek, a hovoří se o referenčním přípravku. Platná tuzemská právní úprava uvádí (ustanovení § 27 odst. 5 zákona o léčivech): „Pokud biologický léčivý přípravek, který je podobný referenčnímu biologickému léčivému přípravku, nesplňuje podmínky vymezení generika, zejména kvůli rozdílům v surovinách nebo rozdílům v postupech výroby takového biologického léčivého přípravku a referenčního biologického léčivého přípravku, musí být předloženy výsledky příslušných předklinických zkoušek nebo klinických hodnocení týkající se těchto podmínek. Výsledky jiných předklinických zkoušek a klinických hodnocení obsažených v registrační dokumentaci referenčního biologického léčivého přípravku se nepředkládají. Prováděcí právní předpis stanoví rozsah doplňujících údajů, které je třeba předložit. Tyto údaje musí být v souladu se souvisejícími pokyny Komise a agentury.“
Z hlediska evropského práva jsou biologická léčiva upravena nařízením Evropského parlamentu a Rady (ES) č. 726/2004 a směrnicí č. 2001/83/ES. V prosinci 2014 byla přijata novelizace Evropské směrnice [9] k podobným biologickým léčivům.
Nebiologická komplexní léčiva
V případě tzv. nebiologických komplexních léčiv (non‑biological complex drugs, NBCDs) je situace složitější, neboť tato léčiva z pohledu tuzemského práva neexistují. Je však zřejmé, že by se nemohlo jednat o generika (zcela odlišný způsob důkazů o ekvivalenci) nebo o biosimilars (zcela odlišný způsob výroby).
Uvedení na trh
Generika mohou být uváděna na trh po uplynutí patentové ochrany, případně po uplynutí lhůty ochrany dat. Velmi zjednodušeně řečeno, pro jejich vstup na trh obvykle dostačuje (vedle splnění řady různých administrativních povinností) studie prokazující bioekvivalenci. Taková studie se většinou provádí na zdravých dobrovolnících, přičemž obvykle postačuje soubor čítající 24 zdravých dobrovolníků. Generikum tak může vstoupit na trh, aniž by je užíval jediný pacient. Generika jsou registrována buď prostřednictvím národní registrace nebo procedurou vzájemného uznávání (mutual recognition procedure, MRP) či postupem decentralizované registrace, v některých případech připadá v úvahu i centralizovaná registrace platná pro celou EU. Může tak docházet a dochází tak k tomu, že část léčivých přípravků obsahujících léčivou látku A má odlišný text Souhrnu údajů o přípravku než jiná část léčivých přípravků obsahujících tutéž léčivou látku A.
Takzvaná biosimilars mohou být též uváděna na trh až po uplynutí patentové ochrany, případně po uplynutí lhůty ochrany dat. Tyto přípravky jsou registrovány vždy centrálním procesem registrace platným pro celou EU.
Porovnání některých parametrů podle typu léčiva je uvedeno v tab. 1. Nejnákladnější je v současnosti vývoj originálního léčivého přípravku, kde nákla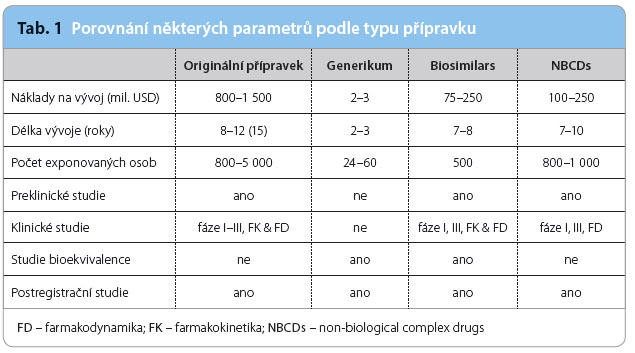 dy obvykle přesahují částku 1 mld. $, vývoj trvá deset let i déle a v průběhu celého procesu je léčivu obvykle exponováno více než 1 000 osob (zdravých dobrovolníků i pacientů).
dy obvykle přesahují částku 1 mld. $, vývoj trvá deset let i déle a v průběhu celého procesu je léčivu obvykle exponováno více než 1 000 osob (zdravých dobrovolníků i pacientů).
Náklady na vývoj generika jsou naproti tomu výrazně nižší a průměrně dosahují pouze zlomku jednoho procenta ve srovnání s originálním přípravkem a exponováno je obvykle kolem 24–48 zdravých dobrovolníků.
Významně nákladnější jsou jak biosimilars, tak i NBCDs, i když s posledně jmenovanými je poměrně málo zkušeností (vedle glatiramoidů jsou do této skupiny zahrnuta např. některá liposomální léčiva, nízkomolekulární hepariny nebo nanočástice obsahující železo).
Zatímco v případě generik nebo biosimilars se provádějí studie bioekvivalence a jejich výsledky jsou rozhodující pro posouzení zaměnitelnosti, v případě NBCDs takové studie postrádají obvykle smysl a neprovádějí se. Celkový výsledek posouzení biosimilars Evropskou lékovou agenturou (European Medicines Agency, EMA) je relativně obtížně predikovatelný. V poslední době EMA posuzovala biosimilars k infliximabu, a ačkoliv byly předloženy důkazy dokládající dostatečnou podobnost v indikaci revmatoidní artritida a ankylozující spondylitida, byly schváleny i veškeré další indikace, které v tu dobu měl originální infliximab (tedy Crohnova choroba, ulcerózní kolitida, psoriatická artritida a psoriáza). Dá se tedy do budoucna očekávat, že podobný „osud“ může potkat další biosimilars (trastuzumab, adalimumab, etanercept), která bude v budoucnosti EMA posuzovat. Jak bude vypadat proces posuzování NBCDs, je v tuto chvíli velkou neznámou.
Glatiramer acetát
Glatiramer acetát není vyráběn pomocí metod založených na rekombinantních technologiích. Velmi pěkný přehled aktuálních informací o glatiramer acetátu přinesl článek profesora Jana Krejska uveřejněný na stránkách tohoto časopisu v prosinci minulého roku [10].
Syntéza glatiramer acetátu na začátku šedesátých let 20. století ve Weizmannově institutu v Izraeli byla motivována snahou najít léčivo, které by mohlo díky podobnosti složení s myelinovým bazickým proteinem obsaženým v nervových zakončeních vyvolat onemocnění morčat (experimentální autoimunitní encefalitidu) sloužící jako zvířecí model roztroušené sklerózy u lidí [11]. Z tehdy syntetizovaných látek vykázal nejvyšší účinek Kopolymer 1, který obsahoval čtyři aminokyseliny – L‑alanin, L‑lysin, kyselinu L‑glutamovou a L‑tyrosin – v poměru 4,2 : 3,4 : 1,4 : 1,0 s průměrnou molekulovou hmotností v rozmezí 5–9 kDa (a se střední molekulovou hmotností 6,4 kDa). Očekávaný efekt se však nedostavil, získaný produkt označený jako Kopolymer 1 měl totiž účinek zcela opačný, experimentální autoimunitní encefalitidu u morčat nevyvolal, ale v experimentálních pracích bylo prokázáno významné působení protektivní.
Je zřejmé, že syntéza proteinu o molekulové hmotnosti přibližně 1 500 kDa s následným štěpením za vzniku zmíněných polypeptidů o obsahu 45–100 náhodně spojených aminokyselin je proces nesmírně složitý zejména z hlediska reprodukovatelnosti výsledků. Ve vyjádření molárních poměrů s příslušným rozmezím pak jsou zmíněné aminokyseliny obsaženy v tomto poměru: 0,392–0,462, 0,300–0,374, 0,129–0,153 a 0,086–0,100. Z toho je zřejmé, že přísné dodržování výrobního procesu nepostačuje, že je nezbytné též náležité testování, zda vyrobený produkt splňuje definované imunologické parametry [11].
Vzhledem k tomu, že farmakokinetické vlastnosti glatiramer acetátu nejsou dostatečně dobře známy, nemají farmakokinetické studie v zásadě žádný význam, a provedení studie bioekvivalence by tedy bylo zcela neprůkazné k doložení dostatečné míry podobnosti. Ve studii u 17 zdravých dobrovolníků [12] byly po podkožní aplikaci dávky 60 mg glatiramer acetátu detekovatelné plazmatické koncentrace pouze u devíti z nich, nadto rozptyl základních farmakokinetických parametrů byl zcela zásadní: velikost plochy pod křivkou činila 1 644–67 532 ng∙min/ml a výše maximálních plazmatických koncentrací dosahovala 69–605 ng/ml.
V případě glatiramer acetátu jako zástupce NBCDs tedy nelze postupovat stejně jako v případě generik, jejichž registrace „odkazem“ právě využívá výsledků studie bioekvivalence. Registrace glatiramoidů postupem určeným legislativou pro generika založená na předložení studie bioekvivalence tedy není z výše uvedených důvodů možná. Na druhou stranu průkaz terapeutické ekvivalence, případně doplněný o výsledky studií in vitro, by teoreticky mohl k registraci postačit. Přesto však by se podle názoru autora nejednalo o registraci generika, neboť by nemohla být splněna podmínka ustanovení § 25 odst. 4 písm. b) zákona č. 378/2007 Sb., o léčivech, tedy plná kvalitativní a kvantitativní shoda v léčivé látce. Registrace glatiramoidů postupem určeným legislativou pro biologická léčiva či biosimilars též není možná, přičemž hlavním důvodem je skutečnost, že při výrobě nejsou použity metody rekombinantní biotechnologie.
Glatiramer acetát je nejprodávanějším přípravkem používaným v léčbě roztroušené sklerózy s ročními prodeji ve výši 4,2 mld. $ (2013). Nemůže proto být divu, že v nedávné době (16. 4. 2015) byla v USA schválena registrace léčivého přípravku Glatopa® [13], který dle amerického Úřadu pro potraviny a léky (Food and Drug Administration, FDA) obsahuje glatiramer acetát. Výrobce léčivého přípravku Copaxone® však byl již předtím (20. 1. 2015) úspěšný u Nejvyššího soudu, kde (sedmi hlasy ku dvěma) soud aproboval názor společnosti Teva ohledně platnosti příslušného patentu v USA. Přesto byl přípravek Glatopa® uveden na trh v USA ve druhé polovině roku 2015 bezprostředně po uplynutí patentové ochrany Copaxone®.
Je přitom velmi zajímavé, že FDA schválil registraci přípravku Glatopa® jako generika, a to na základě zkoumání shod (s glatiramer acetátem) v genové expresi glatiramer‑responzivních myších T‑lymfocytů jako prostředku hodnocení ekvivalence obou léčiv, přičemž jako kontrola posloužil glatiramoid bez schopnosti ovlivňovat diferenciaci subpopulace buněk Th0 T‑lymfocytů na Th2 subsety. Získané výsledky byly následně statisticky zpracovány tak, aby byly vyloučeny náhodné variace v genových expresích. Jiné důkazy či dokumentace ve vztahu k registraci přípravku Glatopa® nebyly na webových stránkách FDA zveřejněny.
Výsledky odlišného přístupu, založeného na průkazu terapeutické ekvivalence, byly publikovány v říjnu 2015. Jedná se o randomizovanou, dvojitě zaslepenou multicentrickou studii [14] s randomizací v poměru 4,3 : 4,3 : 1, ve které byl pacientům s roztroušenou sklerózou podkožně aplikován zkoumaný přípravek (20 mg), přípravek Copaxone® (20 mg), nebo placebo jedenkrát denně po dobu 9 měsíců, přičemž zkoumaným přípravkem je glatiramoid vyvíjený společností Synthon. Primárním cílem studie bylo sledování počtu lézí vychytávajících gadolinium v průběhu sedmi, osmi a devíti měsíců aplikace, dále pak výsledky zobrazení pomocí magnetické rezonance, roční míra relapsů a skóre EDSS (Expanded Disability Status Scale). Bezpečnost a snášenlivost byly hodnoceny pomocí sledování nežádoucích účinků, zejména reakce v místě vpichu a výsledků laboratorních testů. Celkem bylo randomizováno 794 pacientů, kteří dostávali zkoumaný přípravek (n = 353), přípravek Copaxone® (n = 357), nebo placebo (n = 84). Odhadované průměrné počty lézí vychytávajících gadolinium při podávání zkoumaného přípravku a přípravku Copaxone® byly nižší ve srovnání s placebem, a to průměrně o 0,488 (0,365–0,651 na 95% hladině spolehlivosti; p < 0,001). Rozdíly mezi zkoumaným přípravkem a přípravkem Copaxone® nebyly statisticky významné a výskyt byl průměrně mírně vyšší v případě zkoumaného přípravku, a to 1,095 (0,883–1,360 na 95% hladině spolehlivosti). Výskyt, spektrum a závažnost hlášených nežádoucích účinků, včetně reakce v místě vpichu, nebyly statisticky významně rozdílné.
Nezbytnost použití kombinace různých fyzikálních, chemických a biologických metod k potvrzení podobnosti považuje za potřebné výrobce originálního glatiramer acetátu [15]. Důvodem bylo jeho zjištění, že při vcelku dobré shodě v analytických i základních biologických testech ukázaly pokročilé metody, např. celogenomové profilování exprese, při zkoumání glatiramoidů značné rozdíly ve výsledcích řady testů. Na podstatné rozdíly právě v genové expresi přitom ukázal výrobce originálního glatirameru [16] již v minulosti, jednalo se o glatiramoidy z jihoamerického nebo středoamerického kontinentu nebo z Číny. Jakkoliv byly v některých testech odlišnosti značné, nejednalo se o projevy vážné a neočekávané toxicity jako v případě protirameru [17].
Diskuse
S očekávaným budoucím vstupem glatiramoidů na trh v ČR nebo v jiných zemích EU bude spojena celá řada medicínských, ekonomických, právních a etických aspektů. Z hlediska medicínského je zřejmé, že budou‑li k dispozici jednoznačné důkazy jak o účinnosti, tak i o bezpečnosti (včetně případných odchylek z hlediska imunogenicity) prokazující non‑inferioritu, nebude existovat důvod pro odmítání takových NBCDs.
Zvláště v oblasti imunogenicity a rizik plynoucích z nedostatku informací je třeba být mimořádně obezřetný v případě glatiramoidů. Důvody jsou nejméně dva. Prvním je skutečnost, že ze skupiny biologických léčiv je známo, že vznik neutralizačních protilátek vede ke snižování účinnosti u části pacientů. Je přitom zajímavé, že ve skupině biologických léčiv používaných v revmatologii se účinek snižuje v případě revmatoidní artritidy, nikoliv však v případě ankylozující spondylitidy [18]. Druhým důvodem pak může být skutečnost specifických vlastností glatiramoidů, které – na rozdíl od biologických léčiv používaných v revmatologii – mají „základní chemickou strukturu“ alespoň mírně odlišnou v každé šarži a je v současné chvíli zcela neznámo, jakým způsobem budou nemocní na aplikaci glatiramoidů reagovat za 2, 5 nebo 10 let. Hlavním problémem přitom patrně nebudou neutralizační protilátky proti intaktní molekule glatiramoidu, ale proti polypeptidům, které vznikají jeho primárním acidolytickým štěpením. V případě glatiramer acetátu je imunogenicita zcela zásadním problémem, neboť je spojena jak s bezpečností léčby, tak i s její účinností, včetně účinků na molekulární a buněčné úrovni [19].
Z hlediska ekonomického je tuzemský systém regulace cen a úhrad na vstup léčiv ze skupiny NBCDs v zásadě připraven již nyní, neboť podle ustanovení § 39a odst. 5 písm. c) zákona č. 48/1997 Sb., o veřejném zdravotním pojištění, by takový přípravek měl cenu výrobce oproti originálnímu léčivému přípravku sníženu o 15 % a obdobně by byla stanovena i výše jeho úhrady o 15 % méně oproti originálnímu přípravku, a to podle ustanovení § 39c odst. 9 písm. b).
Z právního hlediska pak bude zásadní, jaké budou schváleny indikace a další části Souhrnu údajů o přípravku, z hlediska znění již citovaného zákona o léčivech takový přípravek nemůže být v lékárně obecně zaměnitelný s originálním přípravkem, neboť ustanovení § 83 odst. 2 tohoto zákona vyžaduje splnění podmínky obsahu totožné léčivé látky („… lékárník je oprávněn zaměnit předepsaný léčivý přípravek za jiný léčivý přípravek, který je shodný z hlediska jeho účinnosti a bezpečnosti, obsahuje stejnou léčivou látku se stejnou cestou podání a stejnou lékovou formou“).
Nejsložitějším aspektem bude aspekt etický, a to zejména jeho případné spojení s ekonomickými dopady vstupu podobného přípravku na trh podle zákona o veřejném zdravotním pojištění. Je žádoucí zajistit, aby pacienti léčení určitým konkrétním přípravkem mohli v této léčbě pokračovat bez nutnosti doplácení.
Je velmi obtížné odpovědět na otázku, zda budou přijaty legislativní změny, které by NBCDs zakomponovaly do našeho právního řádu. Spíše bych vycházel ze záporné odpovědi, a proto bude významným úkolem Ministerstva zdravotnictví připravit na vstup NBCDs zejména systém veřejného zdravotního pojištění.
Závěr
Glatiramer acetát je prokazatelně účinné a bezpečné léčivo používané již více než dvacet let v léčbě roztroušení sklerózy [20], jehož patrně jedinou nevýhodou je nutnost každodenní aplikace. I tato relativní nevýhoda bude v brzké době eliminována, neboť úspěšně proběhly kontrolované klinické studie, ve kterých se glatiramer acetát aplikoval v dávce 40 mg 3krát týdně, přičemž byla tato léčba nejen dobře tolerována, ale vykázala lepší účinnost než konvenční dávkování [21].
Glatiramoidy představují zajímavou skupinu léčiv. Některé z již připravených látek jsou v ovlivnění modelových expresí zcela neúčinné, respektive mají opačný efekt (navozují diferenciaci subpopulace Th0 T‑lymfocytů na Th1 či Th17 subsety) a mohou mít problémy z hlediska imunogenicity [22,23], další pak mají sice shodnou účinnost jako glatiramer acetát, avšak se statisticky významnými rozdíly [19].
Po případném vstupu glatiramoidů na trh nelze v žádném případě doporučit „volnou úvahu“ v zaměňování či substituci stávající léčby pacientů s roztroušenou sklerózou. Je dobré si uvědomit, že glatiramer a glatiramoidy ovlivňují expresí více než 1 400 genů [24] a že existuje možnost více než 1036 kombinací aminokyselin L‑alaninu, L‑lysinu, kyseliny L‑glutamové a L‑tyrosinu.
Nejspíš by nebylo od věci inspirovat se experty z Latinské Ameriky [25] a zahájit diskusi o problému NBCDs ještě předtím, než se projeví v plné síle.
Konečné rozhodnutí o tom, jakým přípravkem má být pacient léčen, musí vždy učinit lékař, jak ostatně uvádí EMA (pro biosimilars): „Since biosimilar and biological reference products are not identical, the decision to treat a patients with a reference product or biosimilar medicine should be taken following the opinion of a qualifed health professional.“ Jde jen o to, aby měl pro své rozhodování co nejvíce, pokud možno, objektivních informací.
Seznam použité literatury
- [1] Mayer O. Originální lék versus generikum. Interni Med 2012; 14: 396–398.
- [2] Suchopár J. Originální lék versus generikum. Alergie 2014; 2: 81–85.
- [3] Toverud EL, Hartmann K, Håkonsen H. A Systematic Review of Physicians‘ and Pharmacists‘ Perspectives on Generic Drug Use: What are the Global Challenges? Appl Health Econ Health Policy 2015; 13 (Suppl 1): S35–S45.
- [4] Fuksa L, Vocelka M, Vytrisalova M. The impact of changes in national prescribing conditions for statins on their public expenditure and utilization in the Czech Republic 1997–2013. Health Policy 2015; 119: 1255–1264.
- [5] Gagne JJ, Kesselheim AS, Choudhry NK, et al. Comparative effectiveness of generic versus brand name antiepileptic medications. Epilepsy Behav 2015; 52: 14–18.
- [6] Fitzgerald CL, Jacobson MP. Generic substitution of levetiracetam resulting in increased incidence of breakthrough seizures. Ann Pharmacother 2011; 45: e27.
- [7] Erickson SC, Le L, Ramsey SD, et al. Clinical and pharmacy utilization outcomes with brand to generic antiepileptic switches in patients with epilepsy. Epilepsia 2011; 52: 1365–1371.
- [8] Generics and Biosimilars Initiative Journal, dostupné na http://gabi journal.net/gabi journal/about gabi journal (navštíveno 30. 10. 2015).
- [9] European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology derived proteins as active substance: non clinical and clinical issues. EMEA/CHMP/BMWP/42832/2005 Rev1. 2014.
- [10] Krejsek J. Glatiramer acetát – protizánětlivé a neuroprotektivní mechanismy účinku. Remedia 2014; 24: 464–467.
- [11] Arnon R, Sela M. The Chemistry of the Copaxone Drug, dostupné na https://web.archive.org/web/20030907080548/http://www.weizmann.ac.il/ICS/booklet/1/pdf/copaxon.pdf (navštíveno 30. 10. 2015).
- [12] Messina S, Patti F. The pharmacokinetics of glatiramer acetate for multiple sclerosis treatment. Expert Opin Drug Metab Toxicol 2013; 9: 1349–1359.
- [13] Highlights of prescribing information, Glatopa®, dostupné na http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/090218Orig1s000lbl.pdf (navštíveno 30. 10. 2015).
- [14] Cohen J, Belova A, Selmaj K, et al.; Glatiramer Acetate Clinical Trial to Assess Equivalence With Copaxone (GATE) Study Group. Equivalence of Generic Glatiramer Acetate in Multiple Sclerosis: A Randomized Clinical Trial. JAMA Neurol 2015: Oct 12: 1–9, doi: 10.1001/jamaneurol.2015.2154 [Epub ahead of print].
- [15] Komlosh A, Hasson T, Wells Knecht K, et al. Comparison of physicochemical, biological and genomic characteristics of differently manufactured glatiramoids to ensure MS patient safety. ECTRIMS Congress 2015, Abstr. EP1319, ostupné na http://onlinelibrary.ectrims congress.eu/ectrims/2015/31st/115149arthur.komlosh.comparison.of.physicochemical.biological.and.genomic.html?f=m2.
- [16] Conner J. Glatiramer acetate and therapeutic peptide vaccines for multiple sclerosis. J Autoimmun Cell Resp 2014; 1: 3, dostupné na http://www.hoajonline.com/journals/pdf/2054 989X 1 3.pdf.
- [17] Ramot Y, Rosenstock M, Klinger E, et al. Comparative long term preclinical safety evaluation of two glatiramoid compounds (glatiramer Acetate, Copaxone(R), and TV 5010, protiramer) in rats and monkeys. Toxicol Pathol 2012; 40: 40–54.
- [18] Maneiro JR, Salgado E, Gomez Reino JJ. Immunogenicity of monoclonal antibodies against tumor necrosis factor used in chronic immune mediated Inflammatory condi-tions: systematic review and meta analysis. JAMA Intern Med 2013; 173: 1416–1428.
- [19] Varkony H, Weinstein V, Klinger E, et al. The glatiramoid class of immunomodulator drugs. Expert Opin Pharmacother 2009; 10: 657–668.
- [20] Boster AL, Ford CC, Neudorfer O, Gilgun Sherki Y. Glatiramer acetate: long term safety and efficacy in relapsing remitting multiple sclerosis. Expert Rev Neurother 2015; 15: 575–586.
- [21] Wolinsky JS, Borresen TE, Dietrich DW, et al.; GLACIER Study Group. GLACIER: An open label, randomized, multicenter study to assess the safety and tolerability of glatiramer acetate 40 mg three times weekly versus 20 mg daily in patients with relapsing remitting multiple sclerosis. Mult Scler Relat Disord 2015; 4: 370–376.
- [22] Towfic F, Funt JM, Fowler KD, et al. Comparing the biological impact of glatiramer acetate with the biological impact of a generic. PLoS One 2014; 9: e83757.
- [23] Kolitz S, Hasson T, Towfic F, et al. Gene expression studies of a human monocyte cell line identify dissimilarities between differently manufactured glatiramoids. Sci Rep 2015; 5: 10191.
- [24] Bakshi S, Chalifa Caspi V, Plaschkes I, et al. Gene expression analysis reveals functional pathways of glatiramer acetate activation. Expert Opin Ther Targets 2013; 17: 351–362.
- [25] Carrá A, Macías Islas MA, Tarulla A, et al. Biological and nonbiological complex drugs for multiple sclerosis in Latin America: regulations and risk management. Expert Rev Neurother 2015; 15: 597–600.