Pompeho nemoc – patogeneze, klinický obraz, diagnostika, aktuální možnosti léčby
Souhrn:
Pompeho nemoc, označovaná také jako glykogenóza typu II, je vzácné autosomálně recesivně dědičné onemocnění způsobené mutací genu pro enzym označovaný jako kyselá α glukosidáza. Manifestace onemocnění může nastat v jakémkoliv věku. V novorozeneckém a kojeneckém věku se Pompeho nemoc projevuje jako typická výrazná svalová hypotonie, kardiomegalie a respirační insuficience až respirační selhání. V pozdějším dětství a v dospělosti se one-mocnění manifestuje zejména progredující svalovou slabostí a respirační nedostatečností. Diagnostika onemocnění je založena v první fázi na zjištění aktivity enzymu v suché krevní kapce. Potvrzení diagnózy se provádí vyšetřením aktivity α glukosidázy v leukocytech. Vyšetření DNA je důležité pro stanovení korelace mezi genotypem a fenotypem a pro detekci přenašečů v rodině. V současné době dostupná substituční enzymatická terapie výrazně zlepšila klinický stav pacientů i jejich kvalitu života. Článek je zaměřen na patogenezi, klinický obraz, diagnostiku, a především na možnosti léčby tohoto onemocnění.
Key words: Pompe disease – α glucosidase deficiency – test of dried blood drops – recombinant α glucosidase.
Summary:
Pompe disease, or glycogen storage disease type II, is a rare autosomal recessive disorder caused by mutation in gene that encodes the enzyme acid α glucosidase. Clinical manifestation can start in childhood as well as in adulthood. Severe muscle weakness, cardiomyopathy, and respiratory insufficiency or failure are typical in affected infants. In adulthood, the illness presents with progressive muscle weakness and respiratory insufficiency. First step in the diagnostic process consists of a screening using dried blood spot test. The diagnosis in being confirmed by measurement of α glucosidase activity in leukocytes. DNA testing is important for correlation between the genotype and phenotype as well as for detection of carriers within the family. Treatment with recombinant α glucosidase improves the clinical status of these patients and the quality of their lives. This review is focused on the pathogenesis, presentation, diagnostic process, and treatment options.
Úvod
Pompeho nemoc, označovaná také jako glykogenóza typu II nebo deficit kyselé maltázy, je vzácné autosomálně recesivně dědičné onemocnění, které se projevuje progredující svalovou slabostí. Poprvé byla popsána v roce 1932 nizozemským patologem Johannesem C. Pompem u kojence, který zemřel na idiopatickou srdeční hypertrofii a byla u něj zjištěna masivní akumulace glykogenu v mnoha tkáních, převážně v kosterním svalstvu a v srdci [1].
V roce 1963 byla objevena souvislost tohoto onemocnění s dědičným nedostatkem enzymu kyselé α‑glukosidázy (acid α‑glucosidase, GAA), který je zodpovědný za štěpení glykogenu v buňkách. Výsledkem je intralysosomální akumulace glykogenu, která vede k postupné ztrátě svalových funkcí. Incidence Pompeho choroby se odhaduje na 1 : 40 000–150 000 [2], v ČR by tedy mělo být 74–250 pacientů s touto diagnózou.
Patogeneze
Pompeho nemoc je podmíněna mutacemi genu pro GAA na chromosomu 17q25,3‑q25,3, kterých je v současné době popsáno téměř 300 [3]. Výsledkem těchto mutací je nedostatečná tvorba enzymu GAA. Myocyty, ve kterých je nedostatek enzymu, nejsou schopné štěpit glykogen. Ten se hromadí v lysosomech svalových buněk. Předpokládá se, že akumulace glykogenu vyvolává proces autofagie. Zvýšení počtu lysosomů a autofagosomů může způsobit svalovou slabost mechanickým porušením kontraktilního aparátu svalových vláken [4]. Glykogen se hromadí v lysosomech všech tkání, ve velké míře pak v příčně pruhovaném svalstvu a v myokardu. Akumulace v myokardu bývá nápadná zejména u infantilní formy nemoci a způsobuje hypertrofickou kardiomyopatii, která je pro tuto formu typická. Mezi další orgány a tkáně, u kterých můžeme hromadění glykogenu prokázat (i když méně než v příčně pruhovaném svalstvu a v myokardu), patří játra, ledviny nebo svalovina cév. V CNS nacházíme glykogen ve spinálních gangliích, v předních rozích míšních, v motorických jádrech mozkového kmene a v glii.
Klinický obraz
Příznaky a projevy Pompeho nemoci jsou značně proměnlivé a mohou se objevit v kterémkoli věku od dětství až po dospělost. Rovněž tíže postižení se může u jednotlivých pacientů značně lišit. Podle věku při vzniku onemocnění, závažnosti postižení a podle rychlosti progrese rozlišujeme tři základní formy – infantilní, juvenilní a adultní [5].
Infantilní forma
Jedná se o nejtěžší formu onemocnění. Rozvíjí se brzy po narození a velmi rychle progreduje. Prakticky nulová aktivita GAA vede k výrazné kumulaci glykogenu ve tkáních, zvláště v kosterních svalech a v myokardu [6]. Projevuje se výraznou svalovou slabostí a hypotonií (tzv. obraz floppy infant). Rodiče velmi často udávají potíže s příjmem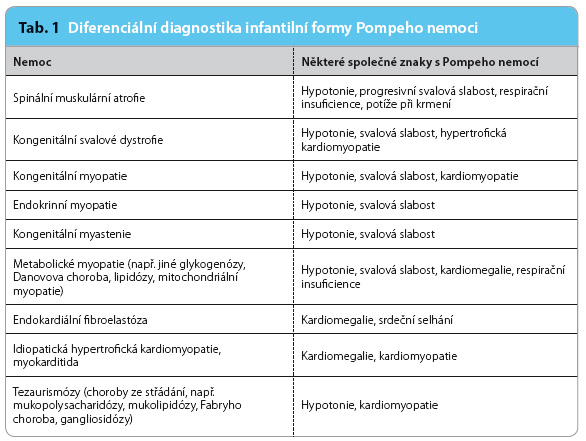 potravy a slabý křik dítěte. Pozorována je tachypnoe a ortopnoe pro slabost respiračního svalstva, časté jsou i recidivující infekce dýchacích cest. Typickým znakem této formy je pak kardiomegalie. V laboratorních odběrech nacházíme výrazně zvýšenou koncentraci kreatinkinázy, a to až desetkrát. Bez léčby většina těchto dětí umírá během prvního roku života v důsledku kardiálního nebo respiračního selhání [7]. Z hlediska diferenciální diagnózy je potřeba odlišit Pompeho nemoc od jiných příčin svalové hypotonie a kardiomyopatie (tab. 1).
potravy a slabý křik dítěte. Pozorována je tachypnoe a ortopnoe pro slabost respiračního svalstva, časté jsou i recidivující infekce dýchacích cest. Typickým znakem této formy je pak kardiomegalie. V laboratorních odběrech nacházíme výrazně zvýšenou koncentraci kreatinkinázy, a to až desetkrát. Bez léčby většina těchto dětí umírá během prvního roku života v důsledku kardiálního nebo respiračního selhání [7]. Z hlediska diferenciální diagnózy je potřeba odlišit Pompeho nemoc od jiných příčin svalové hypotonie a kardiomyopatie (tab. 1).
Juvenilní forma
Juvenilní forma onemocnění má mírnější průběh než forma infantilní. Manifestuje se v průběhu dětství opožděním motorického vývoje, svalovou slabostí zejména v oblasti pletenců a recidivujícími respiračními infekty v důsledku slabosti dýchacího svalstva. Prvním příznakem onemocnění může být i skolióza podmíněná slabostí trupového svalstva. Onemocnění postupně progreduje a bez léčby tito jedinci umírají v časné dospělosti, průměrný věk dožití je 25 let [8].
Adultní forma
První příznaky adultní formy Pompeho nemoci se mohou objevit v časné dospělosti, ale i ve stáří. Průměrný věk nástupu potíží se udává mezi 30. a 40. rokem života [9]. Onemocnění se projevuje progredující svalovou slabostí zejména v oblasti pletenců. Tíže postižení je individuální, ale prakticky u všech pacientů jsou více a dříve postiženy svaly pánevního pletence. I proto patří mezi první přízn aky potíže s chůzí, a to zejména s chůzí do schodů a ze schodů, problémem je chůze na delší vzdálenost nebo schopnost zvednout se z nízké židle. Poměrně častým projevem bývá i únava, intolerance zátěže, myalgie a svalové křeče. V oblasti ramenního pletence bývají postiženy fixátory lopatky způsobující tzv. odstáté lopatky. Slabost trupového svalstva se projevuje potížemi se vstáváním z lehu a časným rozvojem skoliózy. Asi polovina pacientů má při vzniku projevy respirační insuficience. Často se nejprve manifestuje vleže a ve spánku, což vede k příznakům po probuzení, jako jsou bolest hlavy a pocit neosvěžujícího spánku [6]. U řady pacientů dechové potíže progredují až do té míry, že vyžadují neinvazivní nebo i invazivní ventilaci. Bez léčby dochází postupem času k velmi těžké hybné poruše a k dechovému selhání. Koncentrace kreatinkinázy v krvi bývá jen mírně zvýšená. Řada pacientů je pravděpodobně vedena pod jinými diagnózami a jsou neléčeni nebo léčeni nesprávně. Choroby, pod kterými by se mohli tito pacienti eventuálně skrývat, jsou uvedeny v tab. 2.
aky potíže s chůzí, a to zejména s chůzí do schodů a ze schodů, problémem je chůze na delší vzdálenost nebo schopnost zvednout se z nízké židle. Poměrně častým projevem bývá i únava, intolerance zátěže, myalgie a svalové křeče. V oblasti ramenního pletence bývají postiženy fixátory lopatky způsobující tzv. odstáté lopatky. Slabost trupového svalstva se projevuje potížemi se vstáváním z lehu a časným rozvojem skoliózy. Asi polovina pacientů má při vzniku projevy respirační insuficience. Často se nejprve manifestuje vleže a ve spánku, což vede k příznakům po probuzení, jako jsou bolest hlavy a pocit neosvěžujícího spánku [6]. U řady pacientů dechové potíže progredují až do té míry, že vyžadují neinvazivní nebo i invazivní ventilaci. Bez léčby dochází postupem času k velmi těžké hybné poruše a k dechovému selhání. Koncentrace kreatinkinázy v krvi bývá jen mírně zvýšená. Řada pacientů je pravděpodobně vedena pod jinými diagnózami a jsou neléčeni nebo léčeni nesprávně. Choroby, pod kterými by se mohli tito pacienti eventuálně skrývat, jsou uvedeny v tab. 2.
Diagnostika
Při podezření na Pompeho nemoc probíhá diagnostika ve třech úrovních. První úrovní je screeningové vyšetření aktivity GAA pomocí testu suché krevní kapky (dried blood spot, DBS). Vyšetření je jen minimálně invazivní (jedná se o vpich do paty či do špičky prstu) a výsledek je rychle dostupný. Díky testu s akarbózou, která inaktivuje ostatní neutrální glukosidázy, je specificita i senzitivita této metody vysoká. Tento test lze tedy s výhodou použít pro screening velkého počtu vzorků včetně zvažovaného novorozeneckého screeningu Pompeho nemoci.
Definitivní potvrzení diagnózy se provádí vyšetřením aktivity GAA v leukocytech. V minulosti se enzymatické stanovení aktivity GAA provádělo vyšetřením kultivovaných kožních fibroblastů. Odběr těchto vzorků je však poměrně invazivní a trvá přibližně šest týdnů, než jsou výsledky k dispozici. Proto se od této metody ustoupilo a používá se jen výjimečně.
Třetí úroveň diagnostického procesu představuje molekulárněgenetické vyšetření pacienta i rodinných příslušníků. V současné době je popsáno téměř 300 mutací genu GAA, i když ne u všech byla potvrzena patogenita [10]. V minulosti se pro určení obsahu intracelulárního glykogenu a hodnocení aktivity GAA užívala svalová tkáň získaná z biopsie. Avšak vzhledem k tomu, že obsah glykogenu ve svalové tkáni může kolísat, nepřítomnost akumulace glykogenu ve svalové tkáni Pompeho nemoc nevylučuje. Svalová biopsie proto není sama o sobě pro stanovení diagnózy Pompeho nemoci použitelná, z toho důvodu se v současné době v diagnostice Pompeho nemoci nepoužívá.
Léčba
Na konci 20. století a zejména v prvních letech 21. století došlo k výraznému pokroku v léčbě Pompeho nemoci. V roce 1999 byly zahájeny první klinické studie zkoumající účinek enzymatické substituční léčby (enzyme replacement therapy, ERT) s rekombinantní kyselou α‑glukosidázou (rhGAA). V Evropě i ve Spojených státech amerických byla účinná látka registrována v roce 2006 [11]. Léčba spočívá v intravenózním podávání biotechnologicky vyrobené rhGAA v dávce 20 mg/kg ve 14denních intervalech.
Výroba přípravku vychází z nejmodernějších rekombinantních DNA technologií. K výrobě lidského enzymu GAA se využívá buněk vaječníku čínského křečka (Chinese hamster ovary, CHO). Příprava rhGAA začíná genetickou modifikací hostitelských buněk CHO s cílem vytvořit gen lidské GAA. Stručně řečeno, gen lidské GAA se zavede do expresního vektoru a tato konstrukce DNA se pak vkládá do buněk CHO [12]. Geneticky modifikované buňky CHO se posléze replikují s cílem dosáhnout požadované hustoty buněk a podmínek pro optimální produkci enzymu a sekreci do kapalného prostředí. Po získání kultivačního média se pak rekombinantní lidský enzym GAA izoluje pomocí komplexního čisticího procesu, který zahrnuje opakovanou chromatografii a filtraci. Konečný produkt je připraven ve stabilní formě, sterilně filtrován, plněn do lahviček a lyofilizován do práškové formy.
Od zavedení rhGAA do klinické praxe proběhla celá řada studií zabývajících se dlouhodobým účinkem léčby u pacientů s Pompeho nemocí. Tyto studie ukázaly rozdíly v úspěšnosti léčby u jednotlivých forem onemocnění.
U dětí s infantilní formou onemocnění je včasné zahájení léčby nutností, protože tito pacienti bez léčby umírají během prvního roku života. Výsledky klinických studií ukazují, že podávání rekombinantního enzymu redukuje riziko úmrtí až o 99 % [2]. Došlo také k výraznému zlepšení respiračních funkcí (snížení rizika invazivní ventilace o 92 %), ke zmírnění kardiomyopatie i ke zvýšení svalové síly. Léčba nevede k úplnému „vyléčení“ dětí a u mnoha z nich některé příznaky přetrvávají, i když obvykle v mnohem mírnější formě. U některých dětí došlo jen k minimálnímu zlepšení motorických funkcí. Tito pacienti měli však v době zahájení léčby již výrazně omezené pohybové schopnosti. I tyto výsledky zdůrazňují nutnost včasné diagnostiky onemocnění s možností zahájení substituční terapie.
U pozdní formy onemocnění nejsou výsledky substituční terapie tak zásadní. V roce 2014 publikoval Anderson a kol. práci, která se zabývala účinkem léčby u dospělých s Pompeho nemocí. Výzkumu se účastnilo 62 pacientů s Pompeho nemocí, jejichž průměrný věk byl 46,5 roku. Mezi zkoumané ukazatele patřil šestiminutový test chůze (6 minute walk test, 6MWT), zhodnocení svalové síly (Medical Research Council scale, MRC scale) a vyšetření dechových funkcí – usilovné vitální kapacity (forced vital capacity, FVC). Pacienti byli rozděleni do tří skupin. První skupinu tvořili pacienti léčení méně než 12 měsíců, druhou skupinu léčení 12–36 měsíců a poslední skupinu pacienti, kterým byla léčba podávána více než 36 měsíců. U nemocných došlo k částečnému zlepšení ve všech sledovaných ukazatelích v prvních 36 měsících léčby (nejvíce ve skupině 2, tedy u pacientů léčených 12–36 měsíců). Následně se stav pacientů stabilizoval a k dalšímu zlepšení již nedošlo [13]. Podobně jako u infantilní formy i u pozdní formy platí, že čím dříve je substituce rekombinantním enzymem zahájena, tím většího zlepšení klinického stavu můžeme dosáhnout. Přestože je léčba velmi finančně náročná, je v současnosti indikovaná u všech pacientů bez ohledu na věk při stanovení diagnózy onemocnění.
Závěr
Pompeho nemoc je sice poměrně vzácné, ale léčbou ovlivnitelné onemocnění. Jak již bylo uvedeno výše, úspěšnost léčby závisí především na jejím včasném zahájení. To se odvíjí od včasné a správné diagnostiky onemocnění. Vzhledem k tomu, že je v současné době dostupné jednoduché screeningové vyšetření metodou suché krevní kapky, měli bychom toto vyšetření provést u všech pacientů s neobjasněnou svalovou slabostí, zvýšenou hladinou kreatinkinázy či s respirační insuficiencí nejasné etiologie, stejně tak jako u novorozenců/kojenců s hypotonií, svalovou slabostí a s kardiomegalií.
Optimální péče o pacienty s Pompeho nemocí vyžaduje komplexní multidisciplinární přístup. Tým zajišťující tuto péči by se tedy měl skládat z celé řady odborníků, a to jak lékařů (neurolog, kardiolog, pneumolog, ortoped), tak i nelékařů (fyzioterapeut, logoped, psycholog, sociální pracovník).
Seznam použité literatury
- [1] Fukuda T, Roberts A, Plotz PH, et al. Acid alpha glucosidase deficiency (Pompe disease). Curr Neurol Neurosci Rep 2007; 7: 71–77.
- [2] Kishnani PS, Corzo D, Nicolino M, et al. Recombinant human acid α glucosidase: major clinical benefits in infantile onset Pompe disease. Neurology 2007; 68: 99–109.
- [3] Fukuda T, Ewan L, Bauer M, et al. Dysfunction of endocytic and autophagic pathways in lysosomal storage disease. Ann Neurol 2006; 59: 700–708.
- [4] Špalek P. Pompeho choroba – patogenéza, klinický obraz, diagnostika a enzymatická substitučná liečba. Neurol praxi 2009; 10: 44–48.
- [5] Hagemans ML, Winkel LP, Van Doorn PA, et al. Clinical manifestation and natural course of late onset Pompe’s disease in 54 Dutch patients. Brain 2005; 128: 671–677.
- [6] Voháňka S. Pompeho choroba. Medicína po promoci 2011; 12: 80–84.
- [7] Van den Hout HM, Hop W, van Diggelen OP, et al. The natural course of infantile Pompe’s disease: 20 original cases compared with 133 cases from literature. Pediatrics 2003; 112: 332–340.
- [8] Winkel LPE, Hagemans ML, van Doorn PA, et al. The natural course of non classic Pompe‘s disease: a review of 225 cases. J Neurol 2005; 252: 875–884.
- [9] Di Iorio G, Cipullo F, Stromillo L, et al. Adult onset Pompe disease. Acta Myol 2011; 30: 200–202.
- [10] Kroos M, Poponio RJ, van Vliet L, et al. Update of the Pompe disease mutation database with 107 sequence variants and format for severity rating. Hum Mutat 2008; 29: E13–26.
- [11] Schoser B, Laforet P, Kruijshaar ME, et al. 208th ENMC International Workshop: Formation of European Network to develop a European data sharing model and treat-ment guidelines for Pompe disease Naarden, The Netherlands, 26–28 September 2014. Neuromuscul Disord 2015; 25: 674–678.
- [12] Rossi M, Parenti G, Della Casa R, et al. Long term enzyme replacement therapy for Pompe disease with recombinant human alpha glucosidase derived from Chinese hamster ovary cells. J Child Neurol 2007; 22: 565–573.
- [13] Anderson LJ, Henley W, Wyatt KM, et al. Effectiveness of enzyme replacement therapy in adults with late onset Pompe disease: results from the NCS LSD cohort study. J Inherit Metab Dis 2014; 37: 945–952.