Poruchy růstu – možné příčiny a léčba
Růst je specifickou součástí vývoje organismu v dětství. Nadměrný i malý vzrůst je ovlivněn familiárně, mění se při chronické nemoci a v závislosti na psychosociálním prostředí. Za poruchou růstu mohou být hypothalamické a hypofyzární příčiny, abnormity tvorby nebo struktury růstového hormonu a IGF-1, receptorové poruchy, vývojové vady růstové štěrbiny, poruchy nitrobuněčných mechanismů růstu a chromozomální aberace. Pro substituční léčbu je používán rekombinantní růstový hormon (somatropin) a rekombinantní IGF-1 (mecasermin). Většinu kostních dysplazií a poruchy přenosu signálu uvnitř buněk nelze v současnosti léčebně ovlivnit.
Seznam zkratek
ALS – acid labile subunit (kyselá labilní podjednotka); BNP – brain natriuretic peptide (mozkový natriuretický peptid); EMA – European Medicines Agency; FDA – Food and Drug Administration; GH – growth hormone (růstový hormon); GH-R – receptor pro růstový hormon; IGF-1 – insulin-like growth factor 1 (inzulinu podobný růstový faktor 1); IGFBP3 – insulin like growth factor binding protein (vazebný protein pro IGF-1); IUGR – intrauterine growth restriction (intrauterinní růstová restrikce); SDS – skóre směrodatné odchylky
Úvod
Růst j![Graf 1 Růstový graf a pubertogram; volně podle [10] – Krásničanová, et al., 2000. Postnatální růst dětí má čtyři základní části. Rychlý růst do dvou až tří let věku řízený inzulinem a IGF-2, dětský růst do začátku dospívání řízený růstovým hormonem, růstový výšvih v pubertě, na kterém se podílí souhra růstového hormonu a pohlavních hormonů, a po ukončení longitudinálního růstu ještě získání maximální kostní hmoty. Růstové grafy jsou specifické pro různé populace a jsou vztažené na pohlaví. Pubertogram zobrazuje průběh dospívání a vztahuje ho k aktuálnímu růstu.](https://www.remedia.cz/photo-a-29544---.jpg) e součástí fyziologického vývoje dětí. Od narození do dvou až tří let věku pokračuje bouřlivý intrauterinní růst člověka rychlou infantilní fází, po níž následuje pozvolný dětský růst, který je pak znovu akcelerován v době dospívání, a sice souhrou s pohlavními hormony. Na konci pubertálního růstového výšvihu po dosažení definitivní tělesné výšky na růst ještě navazuje vytvoření maximální kostní hmoty, k němuž dochází přibližně po jedenadvacátém roce života (graf 1).
e součástí fyziologického vývoje dětí. Od narození do dvou až tří let věku pokračuje bouřlivý intrauterinní růst člověka rychlou infantilní fází, po níž následuje pozvolný dětský růst, který je pak znovu akcelerován v době dospívání, a sice souhrou s pohlavními hormony. Na konci pubertálního růstového výšvihu po dosažení definitivní tělesné výšky na růst ještě navazuje vytvoření maximální kostní hmoty, k němuž dochází přibližně po jedenadvacátém roce života (graf 1).
Růst citlivě reaguje na chronická onemocnění, hormonální poruchy (Cushingův syndrom, hypotyreóza a tyreotoxikóza), malabsorpci a psychosociální vlivy. Poruchy růstu jsou nejčastějším důvodem vyšetření u dětského endokrinologa. Jedná se převážně o fyziologické varianty opoždění růstu a dospívání nebo o souvislost s chronickým onemocněním či s podáváním léků. Jen malá část dětí je indikována k léčbě růstovým hormonem. V České republice je rekombinantní růstový hormon dostupný od roku 1992, v centrech pro léčbu růstovým hormonem se jím léčí přibližně 1 800 pacientů, převážně dětí. Zcela výjimečně je používán mecasermin – rekombinantní inzulinu podobný růstový faktor 1. Mecasermin je v ČR od roku 2011 podáván třem pacientům. Náhled na regulaci růstu se v posledním desetiletí změnil. Potvrdil tzv. somatomedinovou teorii (Green 1985, LeRoith 2001, Kaplan a Cohen 2007) [1] o tom, že při vzniku růstové poruchy působí nikoli jeden, ale dva proteinové hormony. Zásadní význam má inzulinu podobný růstový faktor 1 (IGF-1), který je stimulován růstovým hormonem. Zatímco růstový hormon vzniká v hypofýze pod vlivem hypothalamického GHRH (growth hormone releasing hormone), IGF-1 se tvoří v játrech a jeho tvorba je současně podmíněna přiměřenou výživou. IGF-1 i růstový hormon vznikají také parakrinně přímo ve tkáních. Tělesný růst stimuluje každý z hormonů samostatně, ale současně dochází k potenciaci většiny jejich účinků. Ve vybraných aspektech, jako je např. vliv na metabolismus cukrů, působí tyto hormony protichůdně.Růstový hormon má pozitivní účinek na tělesný růst, působí anabolicky, zvyšuje inzulinovou rezistenci a utilizaci lipidů, způsobuje zvyšování objemu kostní hmoty, udržuje objem extracelulární tekutiny a stimuluje tvorbu IGF-1 i IGFBP3 (vazebný protein pro IGF-1).
IGF-1 stimuluje svým účinkem na buněčnou diferenciaci a proliferaci růst téměř všech buněk v těle. Má hypoglykemizující – inzulinu podobný účinek, a podobnost s inzulinem je i ve struktuře molekuly. Snižuje hladinu mastných kyselin a proteinů, zvyšuje objem kostní hmoty a urychluje buněčné stárnutí; samotný IGF-1, bez transportního komplexu s vazebnými proteiny, je faktorem onkogeneze. U pacientů s neuropatií má ambivalentní účinek – některé typy postižení může zmírnit, jiné naopak zhoršuje [2].
Hlavní cílovou strukturou působení růstového hormonu a IGF-1 u savců je růstová ploténka kostí. Obecně platí, že poškozená či abnormální růstová ploténka reaguje na růstový hormon a IGF-1 málo nebo vůbec ne. Léčba kostních a chrupavčitých dysplazií (osteochondrodysplazií) a poruch vývoje kosti (osteogenesis imperfecta) je proto neúčinná. Při podávání růstového hormonu rostou části, které nejsou postiženy, zatímco místa s poruchou zůstávají neovlivněná. Tak se prohlubuje disproporcionalita těla. Výjimkou z tohoto pravidla je insuficience SHOX genu, typická pro Turnerův syndrom a mezomelické dysplazie z okruhu Lériho-Weillova syndromu a Langerovy mezomelické dysplazie [3], kde rostou i postižené části kosti.
Poruchy![Graf 2 Bifázická porucha růstu u chlapce s kraniofaryngeomem; podle [10] – Krásničanová, et al., 2000. Po období, kdy chlapec výrazně převyšoval rodinnou růstovou predikci (modré označení „x“ vpravo v grafu), došlo k růstovému selhání. Mezi 7. a 12. rokem vyrostl pouze o 7 cm, tedy růstová rychlost dosahovala 1,4 cm/rok oproti očekávaným 6–8 cm/rok. Růstové selhání je urgentním stavem v pediatrii.](https://www.remedia.cz/photo-a-29545---.jpg) růstu je možné rozdělit na poruchy funkce osy růstový hormon – IGF-1 a na onemocnění mimo tuto osu. Didaktickými kategoriemi jsou nadměrný vzrůst přesahující +2 směrodatné odchylky (SDS) průměrné výšky populace a malý vzrůst (tělesná výška nižší než -2 SDS) [4]. Výjimečně může mít patologie růstu i bifázický charakter, situace je známá u kraniofaryngeomu (graf 2) [5].
růstu je možné rozdělit na poruchy funkce osy růstový hormon – IGF-1 a na onemocnění mimo tuto osu. Didaktickými kategoriemi jsou nadměrný vzrůst přesahující +2 směrodatné odchylky (SDS) průměrné výšky populace a malý vzrůst (tělesná výška nižší než -2 SDS) [4]. Výjimečně může mít patologie růstu i bifázický charakter, situace je známá u kraniofaryngeomu (graf 2) [5].
Poruchy funkce osy růstový hormon – IGF-1
První skupinou onemocnění zasahujících do tvorby růstového hormonu jsou tumory, cysty a hamartomy v oblasti hypothalamu a hypofýzy. Způsobují zvýšenou i sníženou sekreci růstového hormonu. Jejich chirurgická léčba bývá doplňována radioterapií, aplikací lokálních sklerotizačních látek, brachyterapií apod. Výsledky léčby závisí na zkušenosti pracoviště. Typickým příkladem v dětství je kraniofaryngeom, biologicky benigní, ale umístěním závažný nádor Rathkeho výchlipky.
Adenomy hypofýzy produkující růstový hormon jsou v dětství vzácné. Způsobují gigantismus a na hranici dospělosti pak gigantoakromegalii. Léčba může být chirurgická nebo konzervativní – analogy somatostatinu. Ve druhé linii léčby se podává antagonista růstového hormonu pegvisomant, někdy bromokryptin nebo kabergolin. Hypofyzární léze mohou způsobovat izolovanou nebo mnohočetnou hormonální poruchu. K úpravě růstu u pacientů s mnohočetnými deficity je nezbytné zajištění současné normální hladiny ostatních hormonů.Zvláštní příčinou gigantismu nebo akromegalie je aktivační mutace α-podjednotky G-proteinu spojeného s receptory [6]. Stav je označován jako McCuneův-Albrightův syndrom a vyznačuje se pestrou škálou dalších klinických příznaků. Somatotropní buňky jsou na rozdíl od adenomů přítomny v hypofýze difuzně. Užití farmak je léčbou volby.
Druhou velkou skupinu poruch růstu představují poruchy osy růstový hormon IGF-1 s genetickým podkladem. Tyto poruchy vedou k proporčnímu růstu nebo k růstu s nepřiměřeně malou, nebo naopak velkou hlavou, označovanou jako mikrocefalie nebo makrocefalie. Děti s vrozeným deficitem růstového hormonu mají normocefalii, jejich porodní hmotnost i délka se nachází v referenčním rozmezí. Růstový hormon účinkuje od třetího trimestru těhotenství, ale ke klinicky významné poruše růstu dochází až mezi druhým a třetím rokem života dětí při výšce kolem 80 cm. Projeví se jako růstové selhání. Postižení tvorby nebo účinku IGF-1 opožďuje růst již před narozením.Diagnostika deficitu růstového hormonu a deficitu IGF-1 se opírá o stanovení jejich hladin. Pro růstový hormon je to stimulovaná hladina po podání inzulinu, pyridostigminu, klonidinu, argininu nebo glukagonu. Dovoluje vyšetřit endogenní sekreci a posoudit dosažení arbitrární hranice kdykoli, bez ohledu na diurnální rytmus růstového hormonu. Také pro IGF-1 existuje test – hladina IGF-1 se měří po několika dnech podávání růstového hormonu. Proto je test označován jako IGF-1 generační test.
Na zák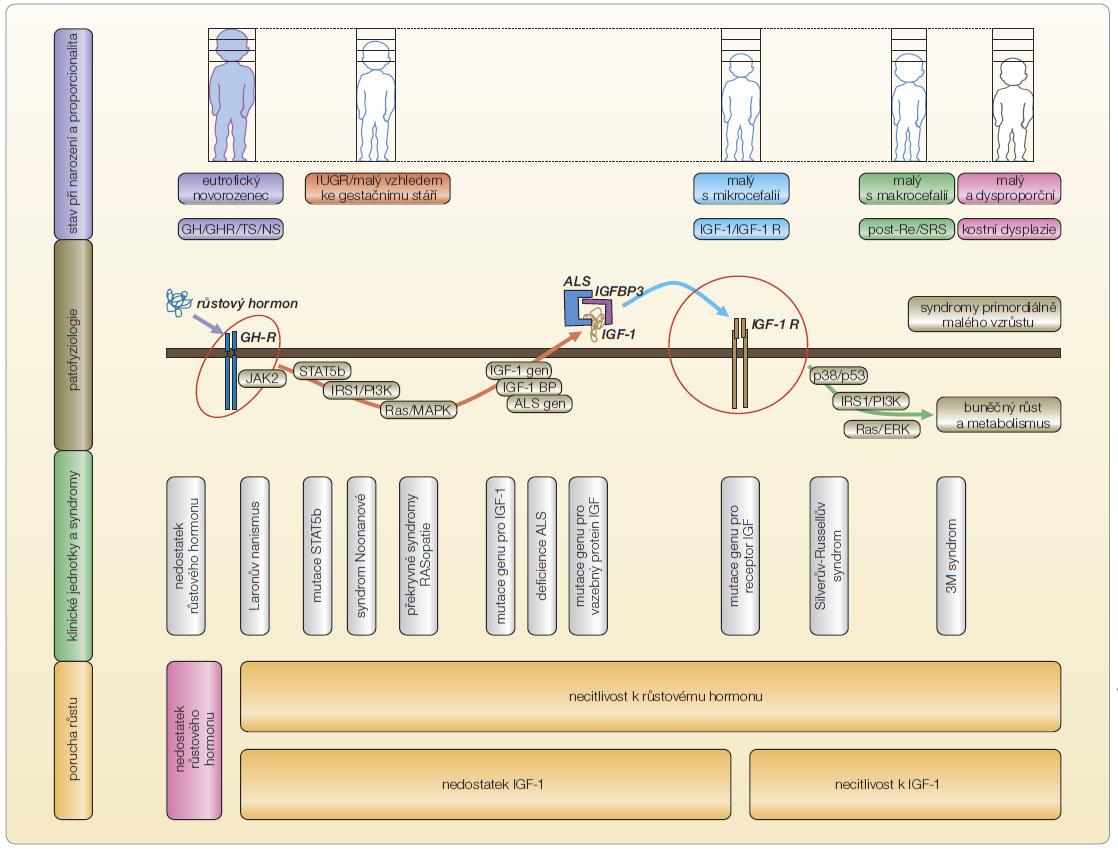 ladě výsledků rozlišujeme deficit růstového hormonu a bioinaktivní růstový hormon. Někdy je zjištěna necitlivost – rezistence k růstovému hormonu; ta je způsobena poruchou receptoru pro růstový hormon, nedostatkem tvorby IGF-1 a poruchou receptoru pro IGF-1 spolu s navazující abnormitou kaskád nitrobuněčných signálů vedoucích k dělení buněk. Poslední situace je označována jako rezistence k IGF-1. V současnosti pro ni neexistuje léčba (obr. 1). Vrozený nedostatek růstového hormonu je izolovaný nebo mnohočetný, a to podle toho, který z genů účastnících se vývoje hypothalamu a hypofýzy je postižen. Některé typy nedostatku růstového hormonu jsou při léčbě doprovázeny tvorbou protilátek. Léčba proto není efektivní [7].
ladě výsledků rozlišujeme deficit růstového hormonu a bioinaktivní růstový hormon. Někdy je zjištěna necitlivost – rezistence k růstovému hormonu; ta je způsobena poruchou receptoru pro růstový hormon, nedostatkem tvorby IGF-1 a poruchou receptoru pro IGF-1 spolu s navazující abnormitou kaskád nitrobuněčných signálů vedoucích k dělení buněk. Poslední situace je označována jako rezistence k IGF-1. V současnosti pro ni neexistuje léčba (obr. 1). Vrozený nedostatek růstového hormonu je izolovaný nebo mnohočetný, a to podle toho, který z genů účastnících se vývoje hypothalamu a hypofýzy je postižen. Některé typy nedostatku růstového hormonu jsou při léčbě doprovázeny tvorbou protilátek. Léčba proto není efektivní [7].
Zvláštní skupinou jsou děti s tzv. idiopatickou malou postavou (ISS – idiopathic short stature). Názory odborníků se liší, ale na tento stav je možné pohlížet jako na nejasně určenou abnormitu s deficitem růstového hormonu na jedné straně a necitlivostí k němu na straně druhé. V roce 2003 schválila FDA (ale ne EMA) podávání růstového hormonu také u těchto dětí. K substituční terapii nedostatku růstového hormonu je používán rekombinantní lidský růstový hormon somatropin (tab. 1). Růstový hormon je připravován biotechnologicky. Je indikován u dětí s deficitem růstového hormonu a u dospělých se závažným stupněm jeho nedostatku, u pacientů s Turnerovým syndromem, s chronickou renální insuficiencí, s Praderovým-Williho syndromem (PWS), u dětí s IUGR bez růstového výšvihu po narození a dětí s deficitem SHOX genu. Podává se subkutánně jedenkrát denně před spaním. Standardní dávkování je 0,025–0,067 mg/kg/den individuálně podle příčiny s následnou titrací podle klinické reakce a hladiny IGF-1. V poslední době se v souvislosti s růstovým hormonem stále více užívá termín „personalizovaná léčba“. Léčba růstovým hormonem zvyšuje ve tkáních přeměnu tyroxinu na trijodtyronin, může vést k benigní intrakraniální hypertenzi, ke vzniku edémů, k lokální reakci v místě aplikace, výjimečně u pacientů s PWS ke spánkové apnoi. Léčba je dlouhodobá. Růstový hormon nezvyšuje riziko vzniku a rekurence
malignit [8].
Biologickou hranicí účinnosti růstového hormonu je jeho receptor. Je-li úplně nebo částečně nefunkční nebo nepracují-li správně kaskády signálů za tímto receptorem, vznikají stavy necitlivosti k růstovému hormonu. Prvním z nich je Laronův nanismus (Laron 1966), způsobený právě mutací receptoru pro růstový hormon [9]. Signál z receptoru pro růstový hormon je nitrobuněčně dále přenášen hlavně kaskádou STAT5b (signal transducers and activators of transcription). Její mutace je další příčinou závažné růstové poruchy, obvykle doprovázené imunodeficitem. Laronův nanismus a poruchy nitrobuněčných kaskád spouštějících transkripci genu pro IGF-1 lze společně označit jako stavy nedostatečné tvorby IGF-1 [1].
Růstový hormon fyziologicky stimuluje nejen tvorbu IGF-1, ale také tvorbu IGFBP3 a ALS (kyselá labilní podjednotka). V krvi se složky spojují ve stabilní ternární komplex a umožňují tak IGF-1 účinkovat na jeho receptory. Také mutace každé ze složek ternárního komplexu vede k poruše růstu. K substituční terapii deficitu IGF-1 se používá rekombinantní lidský IGF-1, který nese název mecasermin. Je schválen k dlouhodobé léčbě růstových poruch dětí a dospívajících od 2 do 18 let se závažným primárním nedostatkem inzulinu podobného růstového faktoru 1 (primární IGFD).
Mecasermin se vyrábí technologií rekombinantní DNA. Podává se subkutánně 2krát denně v době jídla v dávce
0,12 mg/kg u dětí se závažným nedostatkem IGF-1, který je prokázán zvýšenou hladinou růstového hormonu, růstem pod -3 SDS a hladinou IGF-1 nižší než 2,5 percentilu věkově specifické normy. Komplikacemi léčby jsou hypoglykemie, hypertrofie lymfatické t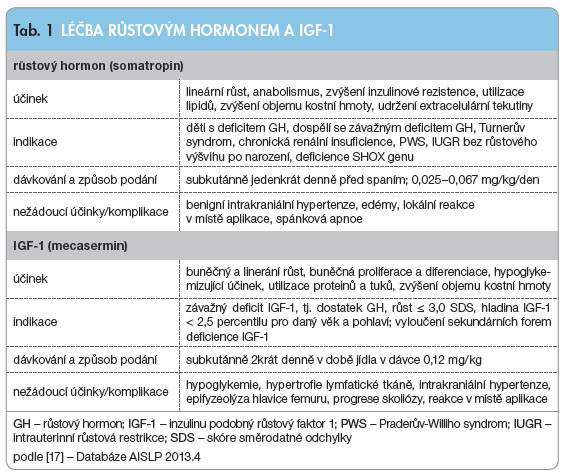 káně (adenoidní vegetace, thymus), intrakraniální hypertenze, epifyzeolýza hlavice femuru, progrese skoliózy, reakce v místě aplikace. Léčebná odpověď je vyhodnocována po jednom roce léčby (tab. 1).
káně (adenoidní vegetace, thymus), intrakraniální hypertenze, epifyzeolýza hlavice femuru, progrese skoliózy, reakce v místě aplikace. Léčebná odpověď je vyhodnocována po jednom roce léčby (tab. 1).
Dalším klíčovým bodem osy růstový hormon – IGF-1 je receptor pro IGF-1
(IGF-R) na buňkách periferních tkání, zvláště na růstové chrupavce (obr. 2). Jeho porucha a následně poruchy postreceptorových mechanismů (kaskády MAPK, PI3K/Akt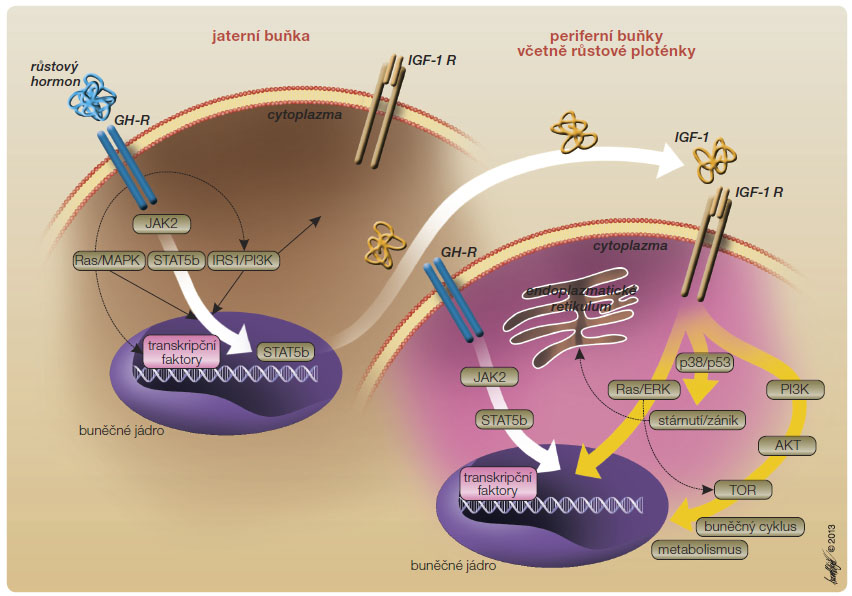 ) jsou příčinou stavů necitlivosti k IGF-1. Farmakologické ovlivnění není v současnosti možné.
) jsou příčinou stavů necitlivosti k IGF-1. Farmakologické ovlivnění není v současnosti možné.
Zvláštní pozici v ovlivnění osy růstový hormon – IGF-1 mají syndromy Noonanové, Silverův-Russellův a 3M syndrom. Klinicky se děti se syndromem Noonanové podobají dívkám s Turnerovým syndromem. Obvykle mívají srdeční vady a je u nich častá autoimunitní tyreoiditida. Defektní nitrobuněčná kaskáda MAPK zde ovlivňuje funkci receptoru pro růstový hormon a kaskádu STAT5b a snižuje tvorbu IGF-1. Silverův-Russellův syndrom je dán epigeneticky. Znamená to, že změny exprese genů nejsou způsobeny mutací DNA, ale změnami chromatinu. Mají souvislost s dědičností v mateřské linii a projevují se až za receptorem pro IGF. Růstová retardace začíná před narozením, děti mají trojúhelníkovitý obličej a klinodaktylii. 3M syndrom je charakterizován extrémně malým proporčním růstem těla a končetin, makrocefalií a nepostiženým intelektem [11]. Nefunguje zde vazba bílkoviny ubikvitinu na degradované proteiny uvnitř buňky. Stav způsobuje rezistenci k růstovému hormonu a/nebo k IGF-1 [12]. Růstovým hormonem jsou léčeny pouze děti se Silverovým-Russelovým syndromem, které spadají do širší skupiny dětí, jež se narodily malé a nedošlo u nich k růstovému výšvihu (IUGR) [13].
Poruchy růstu mimo osu růstový hormon – IGF-1
Ovlivnění syndromů vyvolávajících vysoký vzrůst s poruchou pojiva (Marfanův a Ehlersův-Danlosův syndrom) je pouze symptomatické. Sartany stabilizují kolagen vlivem na transformující růstový faktor TGFβ. Jejich podání je prevencí vzniku cévních aneurysmat [14]. Růst nelze léčebně ovlivnit.
Prenatální a infantilní růst řídí hlavně inzulin a IGF-2. Růstový hormon je v tomto období méně významný. Plody matek s diabetem mellitem, který není správně kompenzován, jsou makrosomické. Důsledná léčba diabetu v těhotenství normalizuje prenatální růst plodu. Jako nadměrně velcí se rodí také novorozenci s Beckwithovým Wiedemannovým syndromem, způsobeným zvýšenou hladinou IGF-2 při duplikaci nebo imprintingu na krátkém raménku 11. chromozomu (11p15).
Sporadicky se vyskytující Sotosův syndrom charakterizuje kombinace vysokého vzrůstu a mentální retardace. Děti mají typický vzhled. Sotosův syndrom je způsobený postižením genu NSD1 (delecí 5q v genu NSD1); funkční protein je ubkviterní a patří do rodiny chromatin modifikujících enzymů. Poškozením stejného genu se vyznačuje také Weaverův syndrom, který je provázen zvýšeným rizikem vzniku malignit [4].
Kostní dysplazie jsou velkou skupinou onemocnění charakterizovaných disproporčním, většinou malým vzrůstem. Fenotyp je obvykle jinak nápadný v novorozeneckém věku, jinak u prepubertálních dětí a jinak u dospělých. V diagnostice je často prospěšné vyšetření proporcionality rodičů. Kostní dysplazií je také deficit SHOX genu, typický pro dívky s Turnerovým syndromem, pro jedince se smíšenou gonadální dysgenezí (45,X0/46,XY) a stavy bodových mutací SHOX genu. SHOX funguje jako transkripční faktor pro natriuretický peptid BNP (brain natriuretic peptide). BNP zásadně ovlivňuje vývoj a růst chrupavky a kosti. Mutace receptoru pro BNP je podkladem pro akromezomelickou dysplazii typu Maroteaux s velkou fenotypovou škálou a růstem na -5 SDS [15].
Skupina konceptu lidí s extrémně malým vzrůstem, tzv. primordiálním nanismem, zahrnuje vzácné syndromy způsobené změnami genů kódujících proteiny buněčné struktury, dělení a apoptózy [16]. Nefunguje signalizace transkripce DNA, funkce centrozomu a mitotického vřeténka, peroxizomů nebo reparace DNA. Klinicky se jedná o proporční jedince se vzrůstem nepřesahujícím jeden metr a s normocefalií nebo mikrocefalií. Patří sem Seckelův syndrom, MOPD (microcephalic osteodysplastic primordial dwarfism), Mulibrey nanismus, Bloomův syndrom a další. Wolcottův-Rallisonův syndrom je charakteristický malým vzrůstem, kostní dysplazií a novorozeneckým diabetem. Příčinou je chybná funkce endoplazmatického retikula a chaperonů, tubulárních bílkovin účastnících se prostorového uspořádání proteinů. Farmakologická léčba kostních dysplazií (s výjimkou deficitu SHOX genu) a lidí s extrémně malým vzrůstem není v současnosti možná.
Podpůrná terapie
Léčba růstovým hormonem je doplňována v případě současného hypogonadotropního hypogonadismu (mnohočetný pituitární deficit) nebo hypergonadotropního hypogonadismu (dysgenetické gonády) substituční hormonální léčbou estrogeny nebo estrogen-gestagenní substitucí u dívek a substitucí testosteronu u chlapců. Vitamin D se podává ve standardním dávkování 600–1 000 jednotek denně.
U syndromů s vysokým vzrůstem je možné ovlivnit definitivní výšku v době dospívání farmakologickým podáním estrogenů u dívek nebo testosteronu u chlapců. Cílem je uzavření kostních štěrbin. Postupy nejsou jednotné, liší se v doporučení věku vhodného pro podání hormonů a v názoru na efekt takové manipulace. Součástí péče o jedince s vysokým vzrůstem je screening hypoglykemií a tumorů.
Další možnosti ve farmakoterapii poruch růstu
V klinickém zkoušení se nachází růstový hormon s prodlouženým účinkem. Inhibitory aromatázy (letrozol, anastrozol) jsou zkoumány u chlapců, u nichž léčba růstovým hormonem začíná v peripubertálním období. Blokáda dospívání prodlužuje terapeutické okno, ve kterém je možné zlepšit růstový zisk, ještě než se uzavřou růstové štěrbiny. U dětí s nedostatkem IGF-1 se studuje vliv mecaserminu podávaného spolu s rekombinantním růstovým hormonem. V současnosti se zdá, že tento nákladný postup zlepšuje výsledky léčby ve srovnání s izolovaným podáním mecaserminu. Kombinace navíc vede k vyvážení hladin IGF-1 a jeho vazebného proteinu. U dívek s Turnerovým syndromem bývá do léčby přidáván oxandrolon, anabolický steroid s nízkou androgenní aktivitou a malým vlivem na růstovou ploténku, který zlepšuje dospělou výšku. U kraniofaryngeomu, jehož buňky jsou epiteliálního původu, je potenciálním přístupem použití retinoidů k zastavení růstu reziduí po operaci.
Závěr
Léčba poruch růstu je v současnosti standardizovaná a soustředěná do center pro léčbu růstovým hormonem. Poruchy, u nichž je indikována léčba mecaserminem, se v ČR objevují zřídka. Největší po-
čet takto postižených se vyskytuje v zemích, kde jsou časté konsanguinní svazky. Porozumění vztahu mezi klinickými projevy a molekulární podstatou poruch růstu pomohlo spolu s dostupností molekulárněgenetického vyšetření zpřesnit a zjednodušit jejich diagnostiku. Léčba poruch růstu pomáhá překonat handicap, který s sebou zvláště malý vzrůst přináší.
Seznam použité literatury
- [1] David A, Hwa W, Metherell LA, et al. Evidence for continuum of genetic, phenotypic, and biochemical abnormalities in children with growth hormone insensitivity. Endocrine Reviews 2011; 32: 472–497.
- [2] Chu Q, Moreland R, Yew NS, et al. Systemic insulin-like growth factor-1 reverses hypoalgesia and improves mobility in mouse model of diabetic peripheral neuropathy. Mol Ther 2008; 16: 1400–1408.
- [3] Zapletalová J, Lebl J, Baxová A, Hirschfeldová K. Defekt v SHOX genu – příčina familiárního malého vzrůstu. Remedia 2010; 20: 313–318.
- [4] Verge CF, Mowat D. Overgrowth. Arch Dis Child 2010; 95: 458–463.
- [5] Müller HL. Childhood craniopharyngioma. Pituitary 2013; 16: 56–67.
- [6] Vortmeyer AO, Gläsker S, Mehta GU, et al. Somatic GNAS mutation cause widespread and difuse pituitary disease in acromegalic patients with McCune-Albright syndrome. J Clin Endocrinol Metab 2012; 97: 2404–2413.
- [7] Pfäffle RW, Blum WF. Understanding the genetics of growth hormone deficiency. Abingdon, TMG Helathcare Communications Ltd, 2000, 87 s. ISBN 1 85113 264 3.
- [8] Rosenfeld RG, Cohen P, Robison LL, et al. Long-term surveillance of growth hormone therapy. J Clin Endocrinol Metab 2012; 97: 68–72.
- [9] Laron Z. Laron syndrome (Primary growth hormone resistance or insensitivity): Personal experience 1958–2003. J Clin Endocrinol Metab 2004; 89: 1031–1044.
- [10] Krásničanová H, Lesný P. Kompendium pediatrické auxologie. Praha, Galén, 2000.
- [11] Neumann D, Hodík K, Šenkeříková M, et al. Speciální diferenciální diagnostika malého vzrůstu. Postgrad Med 2011; 13: 588–597.
- [12] Hanson D, Murray PG, Coulson T, et al. Mutations in CUL7, OBSL1 and CCDC8 in 3-M syndrome lead to disordered growth factor signalling. J Mol Endocrinol 2012; 49: 267–275.
- [13] Toumba M, Albanese A, Azcona C, Standhope R. Effect of long-term growth hormone treatment on final height of children with Russell-Silver syndrome. Horm Res Pediatr 2010; 74: 2012–2017.
- [14] Lin F, Yang X. TGF-beta signaling in aortic aneurysm: another round of controversy. J Genet Genomics 2010; 37: 583–591.
- [15] Marchini A, Häcker H, Marttila T, et al. BNP is a transcriptional target of the short stature homeobox gene SHOX. Hum Mol Gen 2007; 16: 3081–3087.
- [16] Klingseisen A, Jackson AP. Mechanisms and pathways of growth failure primordial dwarfism. Genes Dev 2011; 25: 2011–2024.
- [17] Databáze AISLP 2013.4