Systémová forma juvenilní idiopatické artritidy
Souhrn:
Pojem juvenilní idiopatická artritida (JIA) označuje skupinu chorob, které se v dětském věku prezentují chronickou artritidou a různou mírou mimokloubních projevů. Systémová forma JIA (SJIA) je relativně málo častým, ale zato nejzávažnějším podtypem tohoto onemocnění. Charakterizuje ji přítomnost celkových projevů a často rychle progredující destruktivní polyartritida. Život ohrožující komplikací SJIA je syndrom aktivace makrofágů. Limitujícím faktorem pro prognózu SJIA je včasnost stanovení diagnózy a zahájení adekvátní léčby. Diferenciální diagnóza může být v začátku onemocnění obtížná a zahrnuje jiné zánětlivé choroby i systémové malignity. V terapii se kromě celkově podávaných glukokortikoidů uplatňuje zejména blokáda prozánětlivých cytokinů interleukinu 1 a 6 biologickými přípravky. Jejich podávání obvykle umožní ukončení kortikoterapie a zabrání tak rozvoji dříve často pozorovaných závažných nežádoucích účinků, jako jsou zástava růstu, obezita, hypertenze a katarakta. Problémem zůstává relativně vysoká frekvence relapsů při pokusu o ukončení léčby a řada dětských pacientů si nese různou míru aktivního onemocnění do dospělosti.
Key words: juvenile arthritis – systemic-onset form – diagnosis – therapy – biological therapy.
Summary:
The term juvenile idiopathic arthritis (JIA) describes a group of diseases manifesting themselves during the childhood with chronic arthritis and variable rate of extra‑articular symptoms. The systemic‑onset JIA (SJIA) is the rarest and the most serious form of this disease. It is characterized by the presence of systemic symptoms and often by rapidly progressing destructive polyarthritis. Macrophage activation syndrome is a life threatening complication of SJIA. Early diagnosis, and commencement of appropriate therapy are limiting factors in SJIA prognosis. Differential diagnosis may be challenging at the time of disease onset and include other inflammatory conditions as well as systemic malignancies. Except for systemic glucocorticosteroids, the treatment consists in particular of biological agents blocking the proinflammatory cytokines interleukin 1, and 2. Their administration usually enables the corticotherapy to be stopped and thus prevents the development of serious adverse effects such as growth retardation, obesity, hypertension, and cataract, often seen before. Relatively high relapse rate when attempting to stop the treatment remains an issue and number of paediatric patients carries various level of disease activity to their adulthood.
Úvod
Juvenilní idiopatická artritida (JIA) je podle současně přijatého mezinárodního názvosloví označením pro heterogenní skupinu zánětlivých onemocnění dětského věku projevujících se chronickým zánětem kloubů a variabilní přítomností přidružených příznaků klinických i laboratorních. Svými projevy i prognózou se zásadně liší od revmatoidní artritidy dospělých. I když systémová forma tvoří pouze malý podíl z celkového počtu onemocnění idiopatickou artritidou, závažnost projevů i prognózy ji řadí mezi nejvýznamnější systémová onemocnění u dětí. První klinická charakteristika této choroby byla publikována v roce 1897 anglickým pediatrem J. F. Stillem, podle něhož je označována také jako Stillova nemoc [1].
Obecná charakteristika, epidemiologie, etiologie
Systémová forma juvenilní
idiopatické artritidy (SJIA) je jedinečným dětským onemocněním
zejména kvůli spektru i závažnosti jejích
extraartikulárních projevů, které ji řadí mezi systémové
choroby s variabilně závažnou artritidou. Onemocnění je
definováno pomocí klasifikačních kritérií stanovených
Mezinárodní ligou revmatologických asociací (International League
of Associations for Rheumatology, ILAR), mezi něž patří
přítomnost artritidy a horečky trvající minimálně dva
týdny provázených alespoň jedním z následujících
projevů: typická vyrážka, generalizovaná lymfadenopatie,
zvětšení jater nebo sleziny, serozitida (tab. 1) [2].
Systémová forma juvenilní idiopatické artritidy patří mezi relativně málo časté formy juvenilní artritidy, kde tvoří jen asi 5‒15 % z celkového počtu případů JIA [3,4]. Na rozdíl od ostatních forem JIA, které jsou zpravidla častější u děvčat, se SJIA vyskytuje ve stejné míře u obou pohlaví, nejčastěji ve věku 1‒5 let, i když se může objevit v jakémkoliv věku a vzácně i u dospělých (tzv. Stillova nemoc dospělých).
Etiologie SJIA není objasněna. Rodinný výskyt je vzácný, genetické pozadí nemoci je komplexní a jeví se být odlišné od jiných revmatických chorob včetně ostatních forem JIA. Za významnou je považována asociace s polymorfismy genů kódujících cytokiny vrozené imunitní odpovědi, jako jsou faktor inhibující makrofágy (macrophage inhibiting factor, MIF), tumor nekrotizující faktor alfa (tumor necrosis factor alpha, TNFα), interleukin 6 a interleukin 1 (IL 6, IL 1) a receptor pro IL 1 [5‒8]. Většina těchto polymorfismů má funkční dopad na expresi příslušných cytokinů. Autoprotilátky ani autoreaktivní T lymfocyty nejsou u SJIA přítomny. Zvýšená produkce prozánětlivých cytokinů charakteristických pro aktivaci vrozené imunity (IL 1, IL 6, IL 8, IL 18, MIF, TNF) produkovaných zejména monocyto makrofágovým systémem a neutrofilními leukocyty vysvětluje řadu projevů SJIA [9]. Publikované studie podporují zásadní úlohu nadprodukce IL 6, jehož hodnota je zvýšena v krvi i v synoviální tekutině pacientů se SJIA a koreluje se systémovými projevy onemocnění. Podílí se také na zpomalení růstu, systémové osteoporóze, trombocytóze a anémii – projevech typických pro aktivní onemocnění [10‒13]. Mechanismus vedoucí k nadprodukci IL 1 není plně objasněn, zvažuje se stimulační úloha komplexu transportních membránových proteinů MRP8 a MRP14 produkovaných neutrofily a monocyty/makrofágy [14]. Významná úloha dysregulace mechanismů vrozené imunity vede k chápání SJIA jako autoinflamatorního spíše než autoimunitního onemocnění.
Klinické projevy SJIA
Stav dětí na začátku onemocnění SJIA je obvykle nedobrý ve smyslu celkových projevů ‒ horečky, únavy, bolesti kloubů a svalů, nechutenství s hubnutím, někdy bolesti břicha a na hrudi. Tyto projevy obvykle dominují klinickému obrazu a často předcházejí rozvoji artritidy, někdy o týdny až měsíce.
Horečka u SJIA má typicky intermitentní charakter s jednou nebo dvěma febrilními špičkami nad 39 °C obvykle ráno a/nebo večer s poklesem do normálních až subnormálních hodnot v mezidobí. Pro diagnózu SJIA je nutná přítomnost této kolísavé horečky alespoň dva týdny, z toho ve formálně dokumentované podobě nejméně po dobu tří dnů. V kontrastu s celkovou alterací dítěte při horečce je pro SJIA typické výrazné celkové zlepšení stavu během afebrilního intervalu.
Vyrážka (obr. 1) je obvykle lososově růžová, makulopapulózní,
často s tendencí k centrálnímu výbledu. Obvykle je
prchavá (evanescentní), stěhovavá, objeví se a vybledne bez
rezidua během několika hodin. Typicky provází vzestup teploty
a mizí v době mezi teplotními špičkami. Vzácně může
mít přetrvávající charakter nebo se objevovat nezávisle
na přítomnosti jiných systémových projevů. Její maximum
bývá v oblasti trupu a proximálních částí končetin,
v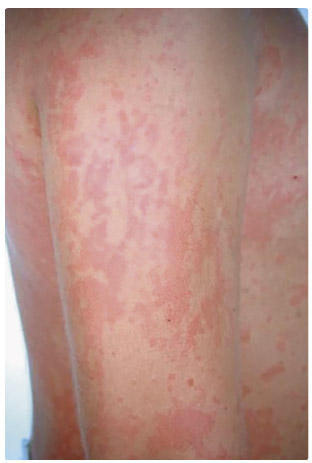 e většině případů nesvědí, i když pruritus je
přítomen až u 10 % pacientů, zejména starších dětí.
Může být přítomen tzv. Koebnerův fenomén, který je označením
pro růžový dermografismus obvykle v podobě pruhovitého
výsevu vyrážky v místech mechanického dráždění např.
oděvem nebo škrábáním.
e většině případů nesvědí, i když pruritus je
přítomen až u 10 % pacientů, zejména starších dětí.
Může být přítomen tzv. Koebnerův fenomén, který je označením
pro růžový dermografismus obvykle v podobě pruhovitého
výsevu vyrážky v místech mechanického dráždění např.
oděvem nebo škrábáním.
Generalizovaná lymfadenopatie a hepatosplenomegalie jsou projevy aktivace retikuloendoteliálního systému. Organomegalie je většinou asymptomatická, i když postižení jater může být provázeno mírným zvýšením aktivity jaterních transamináz, popř. i vzestupem hodnot bilirubinu. Postižení jater je obvykle přechodné, je odrazem systémové aktivity onemocnění, ke chronickému jaternímu onemocnění v rámci SJIA nedochází. Akutní rozvoj progredující jaterní dysfunkce však může být známkou život ohrožující komplikace SJIA, syndromu aktivace makrofágů (viz dále).
Z projevů serozitidy je
nejčastější perikarditida. Je často asymptomatická, může
se ale projevit bolestí na hrudníku, tachykardií
neodpovídající teplotě, třecím šelestem. Její přítomnost je
potvrzena echokardiografickým vyšetřením. Provází obvykle
ostatní projevy systémové aktivity. Pleuritida je vzácnější,
pokud je přítomna, obvykle doprovází perikarditidu. Podezření
na peritonitidu, která patří k nejméně častým
projevům serozitidy, by mělo vzniknout při výraznějších
bolestech břicha.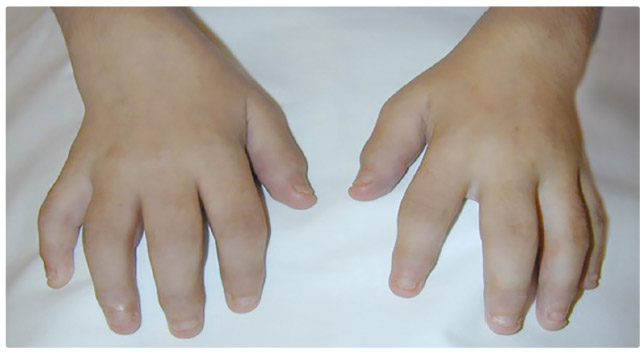
Artritida (obr. 2) může postihovat jakýkoliv počet kloubů, může se objevit současně s ostatními projevy nemoci nebo s různě dlouhým časovým odstupem. U většiny pacientů ale propukne do tří měsíců od první manifestace choroby. Bez objektivně přítomné artritidy nelze diagnózu SJIA potvrdit, i když přítomnost ostatních klinických projevů ji může činit velmi pravděpodobnou. Artritida je obvykle spojena s výraznějšími ranními potížemi jak ve smyslu bolestivosti postižených kloubů, tak funkčního omezení charakteru ranní ztuhlosti. U menších dětí je ranní ztuhlost obtížně zjistitelná, jejich potíže se projevují zvýšenou dráždivostí a odmítáním tělesných aktivit. Často postiženými klouby jsou kolena, zápěstí a kotníky, i když zánět drobných kloubů rukou, krční páteře, čelistních kloubů a kyčlí není výjimkou stejně jako rozvoj obtížně zvládnutelné rychle progredující polyartritidy, která v předbiologické éře obvykle vedla k rozvoji závažné disability. Artritida je často provázena tenosynovitidou zejména v oblasti zápěstí, rukou a nohou, typickým projevem synoviální cysty bicepsu je rozvoj bolestivého otoku paže.
Laboratorní nálezy jsou u SJIA podobně jako u jiných forem tohoto onemocnění nespecifické. V popředí je výrazné zvýšení zánětlivých ukazatelů s vysokou sedimentací erytrocytů (FW), C reaktivního proteinu (CRP) a s neutrofilní leukocytózou. Anémie (obvykle mikrocytární), trombocytóza a snížení koncentrace albuminu odrážejí celkovou míru a trvání zánětlivé aktivity a mohou být výrazné. Zvýšení koncentrace sérového ferritinu je také projevem systémové aktivity nemoci, extrémní hodnoty provázejí syndrom aktivace makrofágů (viz dále). K dalším nespecifickým zánětlivým projevům patří zvýšení sérových koncentrací imunoglobulinů (zejména ve třídách M, G a A) a zvýšení hodnot složek komplementu (zejména C3). S polyklonální zánětlivou aktivací imunitní odpovědi může dojít i k nespecifickému zvýšení titrů protilátek proti řadě antigenů, s nimiž se dítě v minulosti setkalo (např. proti streptokokům, viru Epsteina‒Barrové [EBV], herpes simplex viru apod.). Přítomnost autoprotilátek není pro tuto formu JIA charakteristická. Vysoká zánětlivá aktivita je provázena zvýšením koncentrace cirkulujících prozánětlivých cytokinů i jejich inhibitorů a generalizovanou aktivací cévního endotelu s projevy aktivace koagulační kaskády.
Ze vzácnějších orgánových projevů se může objevit myokarditida provázející perikarditidu, zřídka propuká samostatně. Primární postižení plic patří také ke vzácným projevům SJIA. Nedávno byl popsán soubor 25 pacientů se spektrem plicních příznaků při aktivní SJIA. Plicní arteriální hypertenze byla přítomna u 16 z nich, dvě třetiny nemocných zemřely v průměru za méně než jeden rok [15]. Oční postižení je u SJIA řídké, chronická uveitida se vyskytne u méně než 1 % dětí. Vzácnou komplikací nedostatečně kontrolovaného zánětu u SJIA je sekundární amyloidóza.
Komplikace SJIA
Syndrom aktivace makrofágů
Bezprostředně život ohrožující komplikací SJIA je tzv. syndrom aktivace makrofágů (macrophage activation syndrome, MAS), který se svými projevy podobá hemofagocytující lymfohistiocytóze (HLH). Postihuje až 7 % dětí v průběhu onemocnění SJIA, mortalita se pohybuje kolem 8 % [16,17]. Jeho etiologie je nejasná, podobně jako u HLH je přítomna dysfunkce NK buněk s pravděpodobnou poruchou homeostatických signálů potřebných k odstranění aktivovaných T lymfocytů [18]. Může vzniknout samovolně nebo být vyvolán interkurentní infekcí, zejména herpetickými viry včetně EBV. Obvykle se rozvíjí u dětí s aktivním onemocněním, ve 20 % je zachycen při první manifestaci choroby [19]. Projevuje se rychlým zhoršováním celkového stavu dítěte s trvalou horečkou, zvýrazněním lymfadenopatie a hepatosplenomegalie, v rozvinutém obraze jsou přítomny krvácivé projevy, progredující hepatopatie a neurologická symptomatologie. Nerozpoznaný a neléčený MAS může progredovat do multiorgánového postižení s respirační insuficiencí, selháním ledvin, poruchou vědomí a šokovým stavem. Varujícími laboratorními nálezy jsou nečekaný pokles sedimentace erytrocytů (vlivem konzumpce fibrinogenu v procesu endoteliální aktivace) při vysokém CRP a normálním nebo sníženém počtu trombocytů (popř. i leukocytů a erytrocytů). Dalšími laboratorními parametry je zvýšení hodnoty jaterních transamináz (zejména aspartátaminotransferázy, AST), laktátdehydrogenázy, triglyceridů a extrémně vysoká koncentrace ferritinu v řádu tisíců až desetitisíců (produkován aktivovanými makrofágy), prodloužení aktivovaného parciálního tromboplastinového času (aPTT), pokles koncentrace fibrinogenu a vzestup koncentrace degradačních produktů fibrinu. Diagnóza je klinická, může být potvrzena nálezem hemofagocytujících buněk v aspirátu kostní dřeně, popř. i v jiných orgánech retikuloendoteliálního systému (slezina, játra, lymfatické uzliny), ale až ve 40 % případů rozvinutého MAS nebyla hemofagocytóza nalezena [16].
Vzhledem k život ohrožujícímu stavu u neléčeného MAS je rychlé stanovení diagnózy a zahájení intenzivní terapie nezbytné. Předběžná diagnostická kritéria kombinují klinické projevy s laboratorními nálezy snížení počtu trombocytů pod 262 × 109/l, leukocytů pod 4 × 109/l, vzestup hodnot AST nad 1 µkat/l (59 U/l) a pokles koncentrace fibrinogenu pod 5 g/l [20]. Uvedené hodnoty nepůsobí dramaticky chorobným dojmem, ale je třeba si uvědomit, že dítě s aktivní SJIA bez MAS má typicky neutrofilní leukocytózu, trombocytózu a zvýšenou koncentraci fibrinogenu odpovídající vysoké sedimentaci erytrocytů. Spíš než absolutní číselné hodnoty má význam správné posouzení dynamiky laboratorních nálezů, kdy při klinickém zhoršování stavu dítěte dochází ke „zlepšování“ parametrů nespecifické zánětlivé aktivity.
Sekundární amyloidóza
Sekundární amyloidóza je další vážnou komplikací nekontrolovaného chronického aktivního zánětu u SJIA. Je způsobena tkáňovou depozicí zánětlivého proteinu amyloidu. Její výskyt před érou biologik se pohyboval mezi 5‒7 % [21]. Prakticky u všech dětí bylo v době rozvoje této komplikace přítomno aktivní kloubní onemocnění a vysoké nespecifické laboratorní známky zánětu včetně zvýšení hodnot proteinu akutní fáze sérového amyloidu A. Hlavním úvodním projevem bývá proteinurie, mezi další příznaky patří otoky, hypertenze, bolesti břicha, průjmy. Mortalita je relativně vysoká, hlavní příčinou úmrtí je renální selhání [22]. Diagnóza je stanovena na podkladě nálezu depozit amyloidu v bioptickém materiálu z rektální sliznice, popř. z ledvinné biopsie.
Poruchy růstu a výživy
U dětí s aktivní SJIA je etiologie růstových poruch komplexní. Podobně jako u jiných typů JIA vede chronická artritida k lokálním růstovým změnám postižených kloubů v závislosti na růstové fázi dítěte, délce trvání aktivního zánětu a typu kloubu. U čelistních kloubů, drobných ručních a nožních kloubů dochází častěji k předčasnému uzávěru růstových štěrbin a k rezultujícímu zkrácení těchto struktur (mikromandibula, „tlapkovité“ ruce a nohy). Naopak u kolenního kloubu dochází častěji k přerůstu dlouhých kostí do délky ústícímu při asymetrickém postižení v diskrepanci délky končetin. Celková porucha růstu a neprospívání jsou způsobeny onemocněním samotným, popř. i jeho léčbou. Systémová zánětlivá aktivita u SJIA je spojena s hyperkatabolismem a anorexií s klinickými projevy malnutrice. Zvýšená produkce zánětlivých cytokinů, zejména IL 6, vede k retardaci růstu prostřednictvím omezení produkce inzulinu podobného růstového faktoru I (IGF I). Růstová retardace je dále potencována systémovou kortikoterapií. Léčba celkové poruchy růstu spočívá v první řadě ve snaze o zvládnutí zánětlivé aktivity onemocnění jinými prostředky než systémovou kortikoterapií.
Osteoporóza
Zvýšení kostního obratu způsobené osteoklastickou aktivitou ovlivněnou zejména nadprodukcí IL 6 spolu s demineralizací navozenou kortikoterapií jsou hlavními příčinami generalizované osteoporózy, která zpravidla provází těžší formy SJIA. Nepříjemnou komplikací pokročilé osteoporózy jsou kompresivní fraktury obratlů. V prevenci se uplatňuje kromě intenzivní protizánětlivé terapie suplementace kalcia a vitaminu D3. Základem léčby je ukončení kortikoterapie, v případě patologických fraktur se může uplatnit podání bisfosfonátů [23].
Diagnóza a diferenciální diagnóza
Počáteční klinické projevy jsou společné pro řadu onemocnění dětského věku. Opožděné rozpoznání některých z nich může ohrozit život dítěte. Pokud je u dítěte typické věkové kategorie (předškolák) protrahovaně přítomna charakteristická kolísavá horečka provázená výsevem prchavého exantému s artritidou, lymfadenopatií a hepatosplenomegalií spolu s vysokou sedimentací erytrocytů, leukocytózou a trombocytózou, je diagnóza SJIA velmi pravděpodobná. Tento komplex příznaků je však málokdy přítomen od samého začátku onemocnění. Výjimkou nebývá začátek nemoci provázený pouze horečkou a laboratorními známkami zánětlivé aktivity. Jelikož neexistuje žádné laboratorní vyšetření, které by diagnózu SJIA potvrdilo, a ani žádný jednotlivý projev není pro toto onemocnění specifický, je nutné systematické vyloučení choroby zejména z následujících skupin: infekce, malignity, jiná systémová zánětlivá onemocnění.
Infekce
Systémové infekce jsou nejčastější příčinou horečky neznámého původu u dětí. Kromě běžných infekcí je třeba vyloučit například bakteriální endokarditidu či skrytý absces nebo osteomyelitidu. Pokud je v anamnéze dítěte cesta do oblasti Středomoří, je v diferenciální diagnóze i onemocnění orgánovou formou leishmaniózy. Artritida u virových onemocnění provázených horečkou a exantémem (např. akutní infekce EBV, počáteční stadia hepatitidy typu B) je obvykle krátkého trvání s dobrou odezvou na podání nesteroidních antirevmatik. Obecně horečka u infekčních nemocí je méně předvídatelná, hektičtější, nemá takovou tendenci pravidelně se vracet k normě, stav dítěte se v intervalech mezi teplotními špičkami nezlepšuje tak výrazně jako u SJIA. Artritida u revmatické horečky je akutní a migrující, charakter srdečního postižení je odlišný. Jednorázové zjištění zvýšeného titru antistreptolysinu O tato dvě onemocnění neodliší.
Malignity
Mezi systémové malignity, které mohou svými celkovými i laboratorními projevy napodobovat SJIA, patří zejména akutní hemoblastózy, zejména akutní lymfoblastická leukemie (ALL). Symptomatologie postižení pohybového aparátu je u dětí často jedním z prvních příznaků. Kostní bolest je v začátku leukemie přítomna až u 40 % dětí, může být přítomna i artritida [6‒8]. Bolest je obvykle výraznější než u JIA, může dítě budit v noci. Za její původ je považována expanze kostní dřeně. U artritidy se může vzácně jednat o přímou infiltraci synovie, případně o synoviální reakci na infiltraci periostu či kloubního pouzdra leukemickými buňkami nebo může jít o reaktivní, imunními komplexy indukovanou synovitidu.
Mezi varovné signály, které by měly vést k neodkladnému vyšetření kostní dřeně, patří z klinických projevů zejména nerovnováha objektivního kloubního nálezu a subjektivních potíží dítěte, přítomnost noční bolesti a bolestivosti kostních struktur, nepřítomnost ranní ztuhlosti, výrazná celková symptomatologie (únava, hubnutí, noční poty), horečka jiného než quotidiánního charakteru. Přítomnost lymfadenopatie a hepatosplenomegalie u celkově nemocného dítěte nepomůže rozlišit mezi malignitou a SJIA.
I v nepřítomnosti varujících klinických projevů je třeba hledat laboratorní nálezy, které nejsou typické pro primárně zánětlivé onemocnění. Patří mezi ně zejména:
- neúměrně vysoká sedimentace FW při normálním či sníženém počtu leukocytů a trombocytů (u SJIA zvýšen),
- relativní lymfocytóza (u SJIA neutrofilie),
- neadekvátní stupeň anémie vzhledem k délce trvání potíží (pomalejší rozvoj při chronickém zánětu),
- vysoké hodnoty laktátdehydrogenázy (marker vysokého buněčného obratu).
Nepřítomnost blastů v periferii je v začátku ALL častá a leukemii nevylučuje. Prostý RTG snímek symptomatické oblasti bývá v počátku SJIA bez výrazných změn na skeletu, může být patrna periartikulární osteopenie. U 88 % dětí s leukemií jsou zachycena projasnění v metafýzách dlouhých kostí charakteru Vogtových‒Batyho proužků. Kortikální a periostální léze jsou přítomny v 50 % stejně jako osteolytická ložiska. Významná generalizovaná osteopenie je přítomna prakticky ve všech případech. Vyšetření kostní dřeně vede obvykle ke správné diagnóze. Bohužel v začátku onemocnění leukemií může být v odebraném vzorku dřeně normální nález. V takovém případě je při trvajícím klinickém podezření vhodné pokud možno nepodávat celkově kortikosteroidy a toto vyšetření s nedlouhým časovým odstupem opakovat [9,10].
Diseminovaný neuroblastom se také může projevit celkovými příznaky, kloubními potížemi a infiltrací uzlin. Nepřiměřeně vysoká bolestivost končetiny je varujícím signálem. Pečlivé zhodnocení RTG snímků postižené oblasti a kostní scintigrafie, monitorování krevního tlaku a odpad metabolitů katecholaminů v moči (někdy přítomna endokrinní aktivita), hledání primárního ložiska v oblasti nadledvin a retroperitonea a vzácněji v zadním mediastinu by měly být vždy součástí diferenciálně diagnostického postupu zejména u dětí nejnižší věkové kategorie s neobjasněnou artropatií a celkovými projevy onemocnění.
Výše uvedené klinické projevy v začátku leukemie mohou provázet i řadu jiných maligních onemocnění. Artritida může být vzácně úvodním projevem lymfomů, jejichž diagnóza je při absenci dřeňové infiltrace ještě podstatně obtížnější než u akutní leukemie. Generalizovaná lymfadenopatie nebo přítomnost atypicky lokalizovaných uzlin (supraklavikulárně) u dítěte s muskuloskeletálními potížemi bez jiných známek typických pro SJIA vyžaduje velmi podrobné vyšetření včetně někdy i opakované biopsie uzlin.
Jiná systémová onemocnění
Řada celkových zánětlivých onemocnění se může ve svém začátku podobat SJIA. Patří sem zejména:
- jiná systémová revmatická onemocnění ‒ systémové vaskulitidy (zejména Kawasakiho syndrom, polyarteriitis nodosa), systémový lupus erythematodes;
- jiná systémová zánětlivá onemocnění ‒ chronické střevní záněty (zejména Crohnova choroba);
- autoinflamatorní onemocnění/periodické horečky ‒ periodický syndrom asociovaný s receptorem pro TNF (TRAPS), familiární středomořská horečka, hyper IgD syndrom (HIDS), kryopyrinopatie.
Léčebné strategie
Léčba SJIA je komplexní, založená na multidisciplinárním přístupu k pacientovi a jeho rodině. Jedná se o kombinaci farmakologické léčby, fyzioterapie, edukace a podpůrné léčby psychosomatických a sociálních komplikací onemocnění. Vzhledem ke vzácnosti výskytu SJIA a závažnosti onemocnění je vhodné, aby potvrzení diagnózy a stanovení léčebného plánu proběhlo pod vedením dětského revmatologa na specializovaném pracovišti se zázemím dalších pediatrických specializací i dětské jednotky intenzivní péče. Současná terapeutická doporučení pro SJIA byla formulována v roce 2013 [24]. Volba léku je založena na klinickém obraze (závažnost systémových projevů) a přítomnosti negativních prognostických faktorů, mezi něž patří trvající aktivita onemocnění po šesti měsících od stanovení diagnózy a postižení kyčelních kloubů.
Farmakologická léčba
Léčba systémové formy juvenilní idiopatické artritidy byla v minulosti založena na dlouhodobé kortikoterapii doplněné limitovaně účinnými konvenčními chorobu modifikujícími a imunosupresivními přípravky, které většinou neumožňovaly vysazení kortikosteroidů a zabránění jejich devastujícím nežádoucím účinkům. Děti se SJIA byly dříve mnohem častěji ohroženy invaliditou a podstupovaly aloplastiky destruovaných kloubů. Pokrok v poznání etiopatogenetických mechanismů nemoci umožnil vývoj nové generace přípravků založených na cílené blokádě patologicky aktivovaných imunitních procesů, zejména nadprodukce IL 6 a IL 1. Zavedení biologické léčby do standardních terapeutických algoritmů SJIA přineslo zásadní zlepšení prognózy tohoto onemocnění, i když vysoká frekvence relapsů spojených s ukončováním terapie zůstává nedořešeným problémem.
Nesteroidní antirevmatika
Nesteroidní antirevmatika mají své místo v léčbě horečky a bolesti jako monoterapie zejména v období před potvrzením diagnózy a následně ke krátkodobé symptomatické úlevě při relapsech, kdy jsou podávána v kombinaci s další léčbou. U nás se nejčastěji podává ibuprofen 30‒40 mg/kg/den ve 3‒4 dávkách, popř. indometacin (1,5‒3 mg/kg/den ve třech dávkách) a řada dalších. Účinek i toxicita jsou pro jednotlivé přípravky a u jednotlivých pacientů individuální a je třeba je monitorovat.
Kortikosteroidy
Volba úvodní protizánětlivé léčby závisí na závažnosti onemocnění a na tom, zda převažují projevy systémové, či kloubní. Jejím základem jsou i v současné době kortikosteroidy, ačkoliv do budoucna je pravděpodobné, že by ve vybraných případech mohly být hned v úvodu nemoci nahrazeny biologickou léčbou v rámci tzv. step down přístupu [25]. Intravenózně podávaný metylprednisolon (IVMP) v dávce 20‒30 mg/kg/den, maximálně 1 g, obvykle ve třech po sobě následujících dnech má obvykle rychlý, ale pouze přechodný efekt a musí být následován podáváním nižší dávky perorálního prednisonu (0,5‒1 mg/kg/den či ekvivalentní dávka IVMP) jednou či dvakrát denně podle závažnosti stavu dítěte. Obecně platí, že dělené dávky mají vyšší protizánětlivý účinek, ale také vyšší toxicitu, kvůli níž často volíme jednorázovou dávku v ranních hodinách. U dětí v závažném celkovém stavu někdy dáváme přednost nitrožilní aplikaci i těchto nižších dávek kortikosteroidu z obavy před možným nedokonalým vstřebáváním perorálně podaného přípravku. Pulzní terapie IVMP se uplatňuje nejen při závažných celkových a kloubních projevech, ale i v případě orgánových manifestací, jako je perikarditida, nebo při komplikacích charakteru MAS. U méně závažných stavů lze kortikoterapii zahájit perorálně vyššími denními dávkami prednisonu (1‒2 mg/kg/den). Toxicita kortikosteroidů je vysoce individuální jak z hlediska nástupu jejích projevů, tak s ohledem na dávku přípravku. Obecným doporučením je v případě příznivého terapeutického účinku dávky kortikosteroidů progresivně snižovat během 1‒2 měsíců na dávku 0,2 mg/kg/den a méně a postupně podávání těchto léčiv do šesti měsíců od začátku onemocnění ukončit. Intraartikulární podání depotních kortikosteroidů (triamcinolon hexacetonid) je alternativou při převažujících kloubních projevech či při jejich relapsech, ale obecně je u SJIA méně účinné než u jiných podtypů JIA [26].
Chorobu modifikující léky
Konvenční chorobu modifikující léky mají u SJIA menší význam. Účinek metotrexátu (MTX) na artritidu je nižší než u jiných forem JIA, jeho účinnost na systémové projevy nebyla prokázána [27]. V praxi je jeho podávání dětem s polyartritidou při SJIA obvykle zahájeno v úvodu léčby společně s kortikosteroidy v dávce 15 mg/m2/týden parenterálně, pokud nejsou kontraindikace např. v podobě hepatopatie. Účinek MTX je při souběžné kortikoterapii obtížně hodnotitelný. U velké části dětí dojde v průběhu snižování dávky kortikosteroidů ke zhoršení artritidy nebo k návratu systémových projevů, jež vedou k zahájení biologické léčby obvykle do tří měsíců od stanovení diagnózy. V podávání MTX se pokračuje pouze v případě jeho alespoň částečného účinku. U dětí s převažujícími systémovými projevy je možné zahájit biologickou léčbu s kortikosteroidy nebo bez nich hned v začátku onemocnění. Tento postup naznačuje možnost využití tzv. window of opportunity, krátkého intervalu v průběhu nemoci, během nějž může použití cílené blokády IL 1 (ev. IL 6) zabránit rozvoji chronického zánětu a navodit plnou remisi onemocnění do šesti měsíců od zahájení léčby [28,29].
Blokáda IL 6
Zvýšená produkce IL 6 doprovází vzestupy tělesné teploty a systémovou zánětlivou aktivitu, které jsou pro SJIA charakteristické. Tocilizumab je plně humánní monoklonální protilátka proti receptoru pro IL 6 (IL 6R), která se váže na transmembránovou i solubilní formu IL 6R a brání navázání této molekuly a následnému spuštění prozánětlivé signalizace. V České republice je od konce roku 2013 stanovena jeho úhrada i v pediatrické indikaci. Tocilizumab je předepisován k léčbě aktivní SJIA u pacientů ve věku od dvou let, u nichž je stávající léčba (systémové kortikosteroidy, nesteroidní antirevmatika, MTX v případě, že není kontraindikován nebo intolerován) nedostatečná. Může být podáván jako monoterapie nebo v kombinaci s MTX. Aplikuje se v kapací infuzi v dávce závislé na hmotnosti dítěte, u dětí s tělesnou hmotností nižší než 30 kg v dávce 12 mg/kg, při hmotnosti 30 kg a více pak v dávce 8 mg/kg jednou za dva týdny, s možností prodloužení intervalů na čtyři týdny po dosažení remise onemocnění [30]. Obvykle již po prvním podání dochází k ústupu horečky a k rychlé normalizaci laboratorních zánětlivých parametrů. Tocilizumab je tedy účinný v případě systémových i kloubních projevů tohoto závažného onemocnění, je obvykle velmi dobře tolerován a má přijatelný bezpečnostní profil. Zdá se, že terapie tocilizumabem může navodit ústup rentgenových projevů poškození kloubů. Jeho relativním rizikem je zamaskování laboratorních projevů případných infekčních komplikací. U pacienta léčeného tocilizumabem není možné závažnost infekcí posuzovat na základě míry vzestupu nespecifických laboratorních parametrů (CRP, FW). Antibiotická léčba infekce by měla být zahájena po pečlivém klinickém a mikrobiologickém zhodnocení. Z vážnějších nežádoucích účinků byly zaznamenány případy urtiky, angioedému, leukopenie, nekomplikovaná varicela, septická artritida reagující na antibiotika, přechodná hepatopatie. Tocilizumab je zatím jediným přípravkem ovlivňujícím IL 6, který je schválen ke klinickému použití. V současné době probíhá klinické zkoušení dalších léčiv blokujících účinky IL 6, u dětských pacientů se jedná o plně humánní monoklonální protilátku proti receptoru pro IL 6 sarilumab.
Blokáda IL 1
Nejdéle používaným blokátorem IL 1 je anakinra. Je to lidská rekombinantní forma protizánětlivého proteinu IL 1Ra (antagonista receptoru pro IL 1) produkovaná biotechnologií pomocí bakterie Escherichia coli. Vazbou na receptory pro IL 1 na buněčné membráně brání jejich interakci s IL 1 a následnému spuštění buněčné signalizace. Klinicky významný je její krátký biologický poločas (4‒6 h), jehož výhodou je flexibilita léčby, nevýhodou potřeba každodenní aplikace. I když anakinra není zatím schválena pro použití u dětí se SJIA, publikovaná evidence její roli v léčbě tohoto onemocnění podporuje do té míry, že je součástí terapeutických doporučení [24]. Uplatňuje se zejména v případě systémových projevů, její efekt na artritidu se u části pacientů zdá být menší [31]. V současné době řada specializovaných pracovišť zavedla použití anakinry jako léku první volby u dětí hned v začátku onemocnění SJIA, mnohdy ještě před zahájením systémové kortikoterapie [28,29]. Vzhledem ke krátkému plazmatickému poločasu anakinry a rychlému nástupu terapeutického účinku jsou rizika zejména s ohledem na případnou diagnostickou nejistotu v začátku onemocnění (infekce, malignity) považována za minimální. Anakinra se podává jednou denně v dávce 2 mg/kg tělesné hmotnosti v podkožní injekci. Při nedostatečné odpovědi a při těžkých stavech lze dávku několikanásobně zvýšit a podávat ve 2‒3 denních dávkách intravenózně. Farmakokinetické studie ukazují, že děti s hmotností nižší než 10 kg mohou potřebovat vyšší dávkování [32].
Alternativou k podávání anakinry jsou blokátory IL 1 s prodlouženým biologickým poločasem. Canakinumab je plně humánní monoklonální protilátka proti IL 1β, která ho blokuje, aniž by zkříženě reagovala s podobnými molekulami, jako jsou IL 1α nebo IL 1Ra. Canakinumab má na rozdíl od anakinry dlouhý plazmatický poločas 21‒28 dnů, který umožňuje jeho podávání v delších intervalech. Dětem se SJIA se aplikuje jednou za čtyři týdny v dávce 4 mg/kg v podkožní injekci. Výsledky nedávno publikované kontrolované studie u téměř 200 dětí trpících SJIA a aktivními systémovými projevy vedly k jeho evropské registraci v této indikaci [33]. V České republice má canakinumab zatím schválenu úhradu pouze v indikaci autoinflamatorních onemocnění charakteru kryopyrinopatií. Nevýhodou canakinumabu je jeho relativně vysoká cena ve srovnání s jinými biologiky. Pacienti s nedostatečnou odpovědí na léčbu anakinrou mohou příznivě reagovat na canakinumab [34].
Blokáda TNFα a další léčebné možnosti
Blokáda TNFα není u systémové formy tak účinná jako u jiných podtypů JIA, efekt na systémové projevy nebyl prokázán [35]. Etanercept, adalimumab i infliximab se mohou u SJIA uplatnit po odeznění systémových projevů v případě perzistující polyartritidy [24,36]. Někteří pacienti mohou příznivě odpovědět na blokádu kostimulace abataceptem, výjimečně u rezistentních případů v kombinaci s anakinrou [37,38]. Zatím pouze jedna publikovaná práce dokumentuje příznivý účinek B lymfocyty depletující terapie rituximabem u SJIA [39]. K dalším přípravkům, jejichž použití u SJIA mělo rozporuplné výsledky, patří vysokodávkované intravenózní imunoglobuliny nebo talidomid. Autologní transplantace kmenových buněk je rezervována pro nejtěžší pacienty, u nichž selhala klasická protizánětlivá i biologická léčba. Ze 34 transplantovaných dětí s JIA sledovaných v mezinárodním registru jich mělo 30 systémovou formu onemocnění. U více než poloviny z nich bylo po 12‒60 měsících sledování dosaženo plné remise bez další terapie, mortalita u celého souboru však dosáhla celkem 15 % [40].
Prevence a léčba komplikací SJIA
Nedílnou součástí komplexní léčby je prevence osteoporózy (suplementace vápníku a vitaminu D3), v případě patologických fraktur pak její léčba bisfosfonáty. Nejlepší prevencí i léčbou však zůstává co nejrychlejší dosažení inaktivity onemocnění a ukončení kortikoterapie. Léčba MAS komplikujícího SJIA je založena na vysokodávkovaném pulzním metylprednisolonu obvykle v kombinaci s cyklosporinem A [41]. Čím dříve v průběhu rozvoje MAS je léčba zahájena, tím větší je naděje na zastavení jeho další progrese. Při nedostatečné odpovědi na tuto terapii se uplatňuje zejména podávání anakinry, mnohdy ve výrazně vyšším než standardně doporučeném dávkování [42,43]. V případě rychle progredujícího onemocnění s multiorgánovými projevy je doporučeno podání kombinace kortikosteroidů, cyklosporinu a etoposidu podle protokolu HLH 2004 [44].
Průběh a prognóza
Podle průběhu je možné rozlišit tři klinické podtypy SJIA: monocyklický (celkový výskyt 11 %), intermitentní (34 %) a perzistující (55 %). Při monocyklickém průběhu je dlouhodobá prognóza příznivá, obvykle dojde k ústupu systémových i kloubních projevů onemocnění do 1‒2 let od jeho začátku. U větší části pacientů je však průběh méně příznivý, spojený s rizikem rozvoje dlouhodobých komplikací vlastní nemoci i její léčby. Systémové projevy se mohou nárazově vracet po dobu několika let, samy o sobě však nebývají příčinou trvalého zneschopnění. Hlavním prognózu limitujícím faktorem je rozvoj destruující polyartritidy. Za nepříznivé prognostické faktory je považováno přetrvávání systémových projevů a trombocytóza s počtem trombocytů vyšším než 600 × 109/l po šesti měsících onemocnění, přítomnost perzistující koxitidy a progredující polyartritida. Tento typ průběhu JIA je ze všech jejích klinických forem spojen s nejtěžšími dlouhodobými následky. Po 10 letech sledování mělo aktivní artritidu 48 % pacientů se SJIA. Na celkové úmrtnosti dětí s JIA (0,09 % standardizované mortality v USA) se systémová forma podílí ze dvou třetin [22].
Závěr
Znalost spektra klinických projevů SJIA a adekvátního diferenciálně diagnostického postupu v začátku onemocnění má pro osud nemocného dítěte nesmírný význam. Předčasné zahájení kortikoterapie bez vyloučení systémové malignity může oddálit stanovení správné diagnózy a výrazně tím zhoršit prognózu těchto onemocnění. Nerozpoznání akutní komplikace charakteru MAS je spojeno s vysokým rizikem úmrtí. Na druhé straně neadekvátně vedená kortikoterapie je zatížena významnými nežádoucími účinky. Ačkoliv úvodní diagnostika SJIA je široce pediatrickou záležitostí, definitivní určení diagnózy a stanovení vhodného léčebného postupu patří do rukou dětského revmatologa na specializovaném pracovišti.
Seznam použité literatury
- [1] Still JF. The history of paediatrics. The progress of the study of diseases of children up to the end of the XVIIIth century. College of Paediatrics and Child health, UK, 1996.
- [2] Petty RE, Southwood TR, Manners P, et al. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J Rheumatol 2004; 31: 390–392.
- [3] Danner S, Sordet C, Terzic J, et al. Epidemiology of juvenile idiopathic arthritis in Alsace, France. J Rheumatol 2006; 33: 1377–1381.
- [4] Huemer C, Huemer M, Dorner T, et al. Incidence of pediatric rheumatic diseases in a regional population in Austria. J Rheumatol 2001; 28: 2116–2119.
- [5] Ogilvie EM, Fife MS, Thompson SD, et al. The 174G allele of the interleukin 6 gene confers susceptibility to systemic arthritis in children: a multicenter study using simplex and multiplex juvenile idiopathic arthritis families. Arthritis Rheum 2003; 48: 3202–3206.
- [6] Donn RP, Shelley E, Ollier WE, et al. A novel 5’ flanking region polymorphism of macrophage migration inhibitory factor is associated with systemic onset juvenile idiopathic arthritis. Arthritis Rheum 2001; 44: 1782–1785.
- [7] De Benedetti F, Meazza C, Vivarelli M, et al. Functional and prognostic relevance of the 173 polymorphism of the macrophage migration inhibitory factor gene in systemic onset juvenile idiopathic arthritis. Arthritis Rheum 2003; 48: 1398–1407.
- [8] Stock J, Ogilvie EM, Samuel JM, et al. Comprehensive association study of genetic variants in the IL 1 gene family in systemic juvenile idiopathic arthritis. Genes Immun 2008; 9: 349–357.
- [9] Van den Ham HJ, de Jager W, Bijlsma JW, et al. Differential cytokine profiles in juvenile idiopathic arthritis subtypes revealed by cluster analysis. Rheumatology 2009; 48: 899–905.
- [10] Pignatti P, Vivarelli M, Meazza C, et al. Abnormal regulation of interleukin 6 in systemic juvenile idiopathic arthritis. J Rheumatol 2001; 28: 1670–1676.
- [11] De Benedetti F, Alonzi T, Moretta A, et al. Interleukin 6 causes growth impairment in transgenic mice through a decrease in insulin like growth factor I. A model for stunted growth in children with chronic inflammation. J Clin Invest 1997; 99: 643–650.
- [12] De Benedetti F, Rucci N, Del Fattore A, et al. Impaired skeletal development in interleukin 6 transgenic mice: a model for the impact of chronic inflammation on the growing skeletal system. Arthritis Rheum 2006; 54: 3551–3563.
- [13] Cazzola M, Ponchio L, de Benedetti F, et al. Defective iron supply for erythropoiesis and adequate endogenous erythropoietin production in the anemia associated with systemic onset juvenile chronic arthritis. Blood 1996; 87: 4824–4830.
- [14] Holzinger D, Frosch M, Kastrup A, et al. The Toll like receptor 4 agonist MRP8/14 protein complex is a sensitive indicator for disease activity and predicts relapses in systemic onset juvenile idiopathic arthritis. Ann Rheum Dis 2012; 71: 974–980.
- [15] Kimura Y, Weiss JE, Haroldson KL, et al. Pulmonary hypertension and other potentially fatal pulmonary complications in systemic juvenile idiopathic arthritis. Arthritis Care Res 2013; 65: 745–752.
- [16] Minoia F, Davi S, Horne A, et al. Clinical features, treatment and outcome of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis A multinational, multicenter study of 362 patients. Arthritis Rheumatol 2014; 66: 3160–3169.
- [17] Sawhney S, Woo P, Murray KJ. Macrophage activation syndrome: a potentially fatal complication of rheumatic disorders. Arch Dis Child 2001; 85: 421–426.
- [18] Villanueva J, Lee S, Giannini EH, et al. Natural killer cell dysfunction is a distinguishing feature of systemic onset juvenile rheumatoid arthritis and macrophage activation syndrome. Arthritis Res Ther 2005; 7: R30–37.
- [19] Avcin T, Tse SM, Schneider R, et al. Macrophage activation syndrome as the presenting manifestation of rheumatic diseases in childhood. J Pediatr 2006; 148: 683–686.
- [20] Ravelli A, Magni Manzoni S, Pistorio A, et al. Preliminary diagnostic guidelines for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. J Pediatr 2005; 146: 598–604.
- [21] Schnitzer TJ, Ansell BM. Amyloidosis in juvenile chronic polyarthritis. Arthritis Rheum 1977; 20(2Suppl): 245–252.
- [22] David J, Vouyiouka O, Ansell BM, et al. Amyloidosis in juvenile chronic arthritis: a morbidity and mortality study. Clin Exp Rheumatol 1993; 11: 85–90.
- [23] Noguera A, Ros JB, Pavia C, et al. Bisphosphonates, a new treatment for glucocorticoid induced osteoporosis in children. J Pediatr Endocrinol Metab 2003; 16: 529–536.
- [24] Ringold S, Weiss PF, Beukelman T, et al. 2013 update of the 2011 American College of Rheumatology recommendations for the treatment of juvenile idiopathic arthritis: recommendations for the medical therapy of children with systemic juvenile idiopathic arthritis and tuberculosis screening among children receiving biologic medications. Arthritis Rheum 2013; 65: 2499–2512.
- [25] DeWitt EM, Kimura Y, Beukelman T, et al. Consensus treatment plans for new onset systemic juvenile idiopathic arthritis. Arthritis Care Res 2012; 64: 1001–1010.
- [26] Breit W, Frosch M, Meyer U, et al. A subgroup specific evaluation of the efficacy of intraarticular triamcinolone hexacetonide in juvenile chronic arthritis. J Rheumatol 2000; 27: 2696–2702.
- [27] Woo P, Southwood TR, Prieur AM, et al. Randomized, placebo controlled, crossover trial of low dose oral methotrexate in children with extended oligoarticular or systemic arthritis. Arthritis Rheum 2000; 43: 1849–1857.
- [28] Nigrovic PA. Review: is there a window of opportunity for treatment of systemic juvenile idiopathic arthritis? Arthritis Rheumatol 2014; 66: 1405–1413.
- [29] Vastert SJ, de Jager W, Noordman BJ, et al. Effectiveness of first line treatment with recombinant interleukin 1 receptor antagonist in steroid naive patients with new onset systemic juvenile idiopathic arthritis: results of a prospective cohort study. Arthritis Rheumatol 2014; 66: 1034–1043.
- [30] De Benedetti F, Brunner HI, Ruperto N, et al. Randomized trial of tocilizumab in systemic juvenile idiopathic arthritis. N Engl J Med 2012; 367: 2385–2395.
- [31] Gattorno M, Piccini A, Lasiglie D, et al. The pattern of response to anti interleukin 1 treatment distinguishes two subsets of patients with systemic onset juvenile idiopathic arthritis. Arthritis Rheum 2008; 58: 1505–1515.
- [32] Urien S, Bardin C, Bader Meunier B, et al. Anakinra pharmacokinetics in children and adolescents with systemic onset juvenile idiopathic arthritis and autoinflammatory syndromes. BMC Pharmacol Toxicol 2013; 14: 40.
- [33] Ruperto N, Brunner HI, Quartier P, et al. Two randomized trials of canakinumab in systemic juvenile idiopathic arthritis. N Engl J Med 2012, 367: 2396–2406.
- [34] Quartier P, Grom A, Ruperto N, et al. Efficacy of canakinumab in biologic naive versus previously biologic exposed SJIA patients. Ann Rheum Dis 2014; 73(2Suppl): 66.
- [35] Quartier P, Taupin P, Bourdeaut F, et al. Efficacy of etanercept for the treatment of juvenile idiopathic arthritis according to the onset type. Arthritis Rheum 2003; 48: 1093–1101.
- [36] Kimura Y, Pinho P, Walco G, et al. Etanercept treatment in patients with refractory systemic onset juvenile rheumatoid arthritis. J Rheumatol 2005; 32: 935–942.
- [37] Ruperto N, Lovell DJ, Quartier P, et al. Long term safety and efficacy of abatacept in children with juvenile idiopathic arthritis. Arthritis Rheum 2010; 62: 1792–1802.
- [38] Record JL, Beukelman T, Cron RQ. Combination therapy of abatacept and anakinra in children with refractory systemic juvenile idiopathic arthritis: a retrospective case series. J Rheumatol 2011; 38: 180–181.
- [39] Alexeeva EI, Valieva SI, Bzarova TM, et al. Efficacy and safety of repeat courses of rituximab treatment in patients with severe refractory juvenile idiopathic arthritis. Clin Rheumatol 2011; 30: 1163–1172.
- [40] De Kleer IM, Brinkman DMC, Foerster A, et al. Autologous stem cell transplantation for refractory juvenilie idiopathic arthritis: analysis of clinical effects, mortality and transplant related morbidity. Ann Rheum Dis 2004; 63: 1318–1326.
- [41] Ravelli A, de Benedetti F, Viola S, et al. Macrophage activation syndrome in systemic juvenile rheumatoid arthritis successfully treated with cyclosporine. J Pediatr 1996; 128: 275–278.
- [42] Bruck N, Suttorp M, Kabus M, et al. Rapid and sustained remission of systemic juvenile idiopathic arthritis associated macrophage activation syndrome through treatment with anakinra and corticosteroids. J Clin Rheumatol 2011; 17: 23–27.
- [43] Kahn PJ, Cron RQ. Higher dose anakinra is effective in a case of medically refractory macrophage activation syndrome. J Rheumatol 2013; 40: 743–744.
- [44] Henter JI, Horne A, Arico M, et al. HLH 2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007; 48: 124–131.