Terapeutická zaměnitelnost – příklad anagrelidu
Souhrn:
Anagrelid je významným léčivem při terapii pacientů s esenciální trombocytemií. V blízké době lze v souvislosti s ukončením ochrany duševního vlastnictví očekávat vstup generik anagrelidu na tuzemský trh. Generika anagrelidu budou pro lékaře znamenat novinku nejen ve smyslu možnosti širšího výběru farmak, ale především i z hlediska terapeutické zaměnitelnosti. Jednotlivé léčivé přípravky obsahující anagrelid totiž nebudou s největší pravděpodobností zaměnitelné, tedy ani substituovatelné. Důvodem je skutečnost, že generika anagrelidu se patrně budou odkazovat na léčivý přípravek Xagrid, neboť patentová ochrana léčivého přípravku Thromboreductin ještě trvá. Xagrid přitom není bioekvivalentní ani zaměnitelný s Thromboreductinem. Pochopení jednotlivých zákonných ustanovení týkajících se generických léčiv, jejich zaměnitelnosti a právní odpovědnosti lékaře je významné pro minimalizaci rizik pro pacienta i lékaře.
Key words: anagrelide – imatinib – levothyroxine – Thromboreductin – Xagrid – Eltroxin – Letrox – Glivec – Imatinib Pharmagen – original drugs – generics – bioequivalence – therapeutic interchangeability – generic substitution.
Summary:
Anagrelide is an important drug in the treatment of patients with essential thrombocythemia. Soon, generic anagrelide can be expected to enter the local market in connection with the end of intellectual property rights protection. Generic anagrelide will be a novelty for doctors, not only in terms of a broader choice of medicines but also in terms of therapeutic interchangeability. Most likely, individual medicinal products containing anagrelide will not be interchangeable or substitutable. This is because anagrelide generics will probably be referred to as Xagrid as patent protection for Thromboreductin continues. Xagrid is neither bioequivalent, nor interchangeable with Thromboreductin. Understanding the individual legal provisions on generic medicines, their interchangeability and the legal responsibility of the doctor is important to minimize the risks for the patient and the doctor.
Úvod
V podmínkách České republiky existuje již řadu let neobvyklá situace v oblasti registrace přípravků obsahujících léčivou látku anagrelid. Jsou souběžně registrovány léčivé přípravky Xagrid a Thromboreductin, které oba anagrelid obsahují v množství 0,5 mg v jedné tobolce. Léčivý přípravek Xagrid je registrován evropskou registrací a léčivý přípravek Thromboreductin je registrován národní registrací. Zatímco Thromboreductin je dostupný na trhu a lékaři mají s jeho použitím dlouholeté zkušenosti, Xagrid se od doby své registrace neobchoduje a ani nemá stanovenou výši a podmínky úhrady. Jak bude uvedeno dále, jsou tyto dva léčivé přípravky, ačkoliv obsahují stejné množství anagrelidu, terapeuticky nezaměnitelné, protože jednak nejsou bioekvivalentní a jednak nejsou terapeuticky ekvivalentní z důvodu odlišné bezpečnosti. Na konci minulého roku (16. listopadu 2016) vypršela veškerá příslušná práva pro Xagrid a v ČR se tak mohou registrovat tzv. generické anagrelidy odkazující na registrační dokumentaci tohoto léčivého přípravku, zatímco Thromboreductin nadále bude chráněn patentem z roku 2009 a jeho patentová ochrana potrvá do 4. prosince 2029.
Cílem této práce je poskytnout informace, které lze považovat za důležité pro rozhodování lékaře o terapii a jejích případných změnách a pro lepší orientaci v problematice zaměnitelnosti. Vstupem tzv. generických anagrelidů totiž nastane poměrně kuriózní situace. Ta bude spočívat ve skutečnosti, že pro Xagrid budou patrně existovat generika, zatímco pro Thromboreductin generika patrně ještě po poměrně dlouhou dobu existovat nebudou. Generika k léčivému přípravku Xagrid (podobně jako Xagrid sám, jak již bylo uvedeno) nejsou bioekvivalentní ani terapeuticky ekvivalentní k léčivému přípravku Thromboreductin, a to kvůli rozdílné bezpečnosti. Na druhou stranu lze jen obtížně pochybovat o tom, že výše úhrady se nebude v budoucnosti odvíjet od ceny generik anagrelidu.
Budou tak logicky kladeny otázky, zda bude možné jednotlivé léčivé přípravky u konkrétního pacienta zcela volně zaměňovat při preskripci lékařem, zda bude možné oba přípravky na úrovni lékárny substituovat, případně jaké mohou nastat hmotněprávní důsledky takových postupů. Mohou být také kladeny zásadní otázky ekonomické, zejména proti úsporám se obtížně vznášejí argumenty, přičemž šetřit je nepochybně třeba. Stejně tak může být nadnesena otázka, jaký bude postup zdravotních pojišťoven v případě, že lékař předepíše Thromboreductin s tím, že na lékařském předpisu vyznačí „Nezaměňovat“.
Zákonná definice generika
Zákon o léčivech (zákon č. 378/2007 Sb., v platném znění) umožňuje registrovat léčivý přípravek několika způsoby. Hned na úvod je vhodné poznamenat, že problematika registrací je velmi podrobně upravena právem Evropské unie (EU), a to jednak celou řadou směrnic (které jsou transponovány do vnitrostátního práva), jednak rovněž nařízeními, která jsou aplikovatelná přímo.
Základním cílem registračního řízení je zajistit, aby při poskytování zdravotních služeb byly používány pouze přípravky dostatečně účinné, bezpečné a jakostní.
Prvním způsobem je registrace na základě úplné registrační dokumentace (včetně preklinické a klinické dokumentace dokládající účinnost, bezpečnost a příznivý poměr přínosů a rizik). Další možností je registrace generická, u níž se odkazuje na dokumentaci jiného již registrovaného přípravku. Tento postup je obvyklý po uplynutí patentové ochrany a doby ochrany registračních dat. V takovém případě stačí prokázat bioekvivalenci tohoto generika s referenčním léčivým přípravkem, a to příslušnými studiemi biologické dostupnosti.
Zákon o léčivech přitom definuje generikum (v § 25 odst. 4 písm. b) jako léčivý přípravek, který má shodné kvalitativní a kvantitativní složení, pokud jde o léčivé látky, a shodnou lékovou formu s referenčním přípravkem a u kterého byla (s výjimkou případů, kdy lze doložit, že generikum splňuje příslušná kritéria stanovená pokyny Evropské komise a Evropské lékové agentury, EMA) prokázána bioekvivalence s referenčním přípravkem příslušnými studiemi biologické dostupnosti. Různé soli, estery, étery, izomery, směsi izomerů, komplexy nebo deriváty léčivé látky se považují za tutéž léčivou látku, pokud se významně neodlišují vlastnostmi týkajícími se bezpečnosti, popřípadě účinnosti. Různé perorální lékové formy s okamžitým uvolňováním se považují za jednu a tutéž lékovou formu.
Zákon o léčivech přitom nedefinuje originální léčivé přípravky, což je málo známá skutečnost. „Negenerikum“ obvykle nebylo zkoumáno z hlediska bioekvivalence nebo nemá vědecký průkaz umožňující terapeutickou zaměnitelnost.
Jako referenční přípravek může sloužit jak originální přípravek, tak i jiné generikum.
Speciálním případem je registrace biologických léčivých přípravků (biosimilars). Zákon o léčivech (v § 27 odst. 5) stanoví, že pokud biologický léčivý přípravek, který je podobný referenčnímu biologickému léčivému přípravku, nesplňuje podmínky vymezení generika, zejména kvůli rozdílům v surovinách nebo rozdílům v postupech výroby takového biologického léčivého přípravku a referenčního biologického léčivého přípravku, musejí být předloženy výsledky příslušných předklinických zkoušek nebo klinických hodnocení týkající se těchto podmínek. Výsledky jiných předklinických zkoušek a klinických hodnocení obsažených v registrační dokumentaci referenčního biologického léčivého přípravku se nepředkládají. Avšak absence jakékoliv klinické zkušenosti s použitím takového generika může vyvolávat pochybnosti zejména o bezpečnosti, neboť výskyt řady nežádoucích účinků může být závislý na obsahu a množství pomocných látek, případně i na dalších faktorech (např. na velikosti částic léčiva).
Dalšími případy jsou tzv. hybridní registrace, kdy se dokládají pouze určité skutečnosti (např. v případě změn léčivé látky nebo léčivých látek, léčebných indikací, síly, lékové formy nebo cesty podání ve srovnání s referenčním přípravkem je třeba doložit příslušnou preklinickou a klinickou dokumentaci či doplňující údaje poskytující důkazy o účinnosti či bezpečnosti).
Zákon o léčivech rovněž umožňuje (v § 27 odst. 7) registrovat léčivý přípravek na základě tzv. dobře zavedeného léčebného použití (well established use, WEU). U této registrace není nutno předkládat výsledky předklinických zkoušek a klinických hodnocení, ale prokazuje se fakt, že léčivé látky daného přípravku mají dobře zavedené léčebné použití v EU po dobu alespoň 10 let s uznanou účinností a přijatelnou úrovní bezpečnosti. V takovém případě se předloží příslušná vědecká literatura. Absence jakéhokoliv klinického použití takového léčivého přípravku však může vyvolávat pochybnosti zejména o bezpečnosti, neboť výskyt řady nežádoucích účinků může být závislý na obsahu a množství pomocných látek, případně i na dalších faktorech.
Zákonná definice terapeutické zaměnitelnosti
Zdravotničtí profesionálové vnímají pojem terapeutické zaměnitelnosti obvykle velmi redukcionisticky ‒ obsahuje li léčivý přípravek totožnou léčivou látku, je automaticky terapeuticky zaměnitelný. Z právního hlediska je však situace odlišná.
Zákonná definice terapeutické zaměnitelnosti není uvedena v zákoně o léčivech, ale v zákoně o veřejném zdravotním pojištění (zákon č. 48/1997 Sb., v platném znění).
Ustanovení § 39c odst. 1 tohoto zákona definuje referenční skupiny jako skupiny léčivých přípravků v zásadě terapeuticky zaměnitelných, s obdobnou nebo blízkou účinností a bezpečností a s obdobným klinickým využitím.
Z této definice vyplývá, že jako léčivé, v zásadě terapeuticky zaměnitelné lze přípravky označit pouze při kumulativním splnění těchto předpokladů:
- jde o léčivé přípravky s obdobnou nebo blízkou účinností,
- jde o léčivé přípravky s obdobnou nebo blízkou bezpečností,
- jde o léčivé přípravky s obdobným klinickým využitím.
V této souvislosti je nutno upozornit, že i když dva léčivé přípravky obsahují stejnou účinnou látku (a dokonce ve stejné síle a lékové formě), nemusí to nutně znamenat, že jsou zaměnitelné. Nezaměnitelnost může být způsobena například zásadně odlišnými terapeutickými indikacemi podle souhrnu údajů o přípravku (SPC).
Může například dojít k tomu, že určitá indikace (určitý způsob použití) je dosud chráněna patentem (či dodatkovým ochranným osvědčením), a proto tato indikace (způsob použití) není uvedena v SPC jiného přípravku. Pak takový přípravek lze v off label indikaci použít pouze za zvláštních podmínek, které jsou popsány dále.
Pokud jde o generikum, z jeho definice podle zákona o léčivech (kdy se prokazuje bioekvivalence ‒ viz výše) vyplývá, že tato situace odlišných terapeutických indikací (či způsobu použití) v platných SPC je jedním z nemnoha případů, kdy referenční a generický přípravek nejsou terapeuticky zaměnitelné.
Terapeutickou zaměnitelnost (z pohledu zákona o veřejném zdravotním pojištění, tedy pro účely stanovení úhrady) posuzuje Státní ústav pro kontrolu léčiv. Zde je vhodné poznamenat, že v praxi zde nastávají mnohdy problematické situace, kdy jsou často posouzeny jako v zásadě terapeuticky zaměnitelné i ty přípravky, které vykazují významné odlišnosti jak v účinnosti, tak v bezpečnosti. Takový postup je odůvodňován tím, že není požadována úplná shoda v účinnosti a bezpečnosti. Navíc je používán argument, že stačí jedna jediná společná indikace, která se označí jako tzv. referenční, a to i za situace, kdy je tato indikace z hlediska klinického využití minoritní. Takový přístup lze považovat z odborného hlediska za značně problematický.
Zcela jinak je řešena započitatelnost do ochranného limitu na doplatky na částečně hrazené léčivé přípravky (podle § 16b zákona o veřejném zdravotním pojištění). Do tohoto ochranného limitu se započítávají doplatky na částečně hrazené přípravky (nebo potraviny pro zvláštní lékařské účely) s obsahem stejné léčivé látky a stejné cesty podání pouze ve výši vypočtené podle doplatku na přípravek nebo potravinu pro zvláštní lékařské účely, jehož doplatek na množstevní jednotku této léčivé látky je nejnižší a u kterého nebylo zjištěno přerušení nebo ukončení dodávání. To neplatí, pokud předepisující lékař na receptu vyznačil, že předepsaný léčivý přípravek nelze nahradit (tedy uvedl „Nezaměňovat“). V takovém případě se do limitu započítává doplatek v plné výši. Do limitu se nezapočítávají doplatky na částečně hrazené přípravky nebo potraviny pro zvláštní lékařské účely obsahující léčivé látky určené k podpůrné nebo doplňkové léčbě (tyto doplatky se započítávají pouze pojištěncům starším 65 let). Započitatelnost do ochranného limitu se tedy vztahuje vždy na přípravky s obsahem stejné léčivé látky a stejné cesty podání, terapeutická zaměnitelnost v tomto ohledu nemá žádný význam. Na druhé straně je však třeba upozornit, že vyznačení „Nezaměňovat“ na receptu má dopad na to, že lékaři se do jeho limitu na léky započítají náklady na takový léčivý přípravek.
Možnost generické substituce
Generická substituce je postup, při němž v lékárně není vydán předepsaný přípravek, ale přípravek jiný, což za definovaných podmínek zákonná úprava umožňuje. Zde je třeba uvést, že tato problematika je řešena jak v zákoně o léčivech, tak v zákoně o veřejném zdravotním pojištění (pro účely úhrady).
Zákon o léčivech stanoví (v § 83 odst. 2), že vyznačí li předepisující lékař na lékařském předpisu, že trvá na vydání předepsaného přípravku, lze vydat pouze tento. V ostatních případech lékárník informuje pacienta o možných alternativách k vydávanému přípravku a s jeho souhlasem je oprávněn zaměnit předepsaný léčivý přípravek za jiný, který je shodný z hlediska své účinnosti a bezpečnosti, obsahuje stejnou léčivou látku se stejnou cestou podání a stejnou lékovou formou. Zvláštním případem je situace, kdy lékárník nemá léčivý přípravek předepsaný lékařem k dispozici a je nezbytné jeho okamžité vydání. V takovém případě vydá jiný přípravek odpovídajících léčebných vlastností, který má k dispozici (§ 83 odst. 3 zákona o léčivech).
Zákon o veřejném zdravotním pojištění stanoví (v § 32 odst. 2), že požádá li pojištěnec o vydání jiného přípravku se stejnou léčivou látkou, se stejnou cestou podání a se stejnou lékovou formou, nahradí jej lékárna jiným léčivým přípravkem s nižším doplatkem, pokud předepisující lékař na receptu nevyznačil, že předepsaný přípravek nelze nahradit.
V této souvislosti je ještě závěrem vhodné upozornit na poučovací povinnost poskytovatelů zdravotních služeb podle zákona o zdravotních službách (zákon č. 372/2011 Sb., v platném znění). Ten stanoví (v § 45 odst. 2 písm. a), že poskytovatel je povinen informovat pacienta o ceně poskytovaných zdravotních služeb nehrazených nebo částečně hrazených z veřejného zdravotního pojištění, a to před jejich poskytnutím, a vystavit účet za uhrazené zdravotní služby, nestanoví li jiný právní předpis jinak.
Zákonné podmínky postupu off label
Zákon o léčivech stanoví (v § 7 odst. 1 písm. b), že osoby zacházející s léčivy jsou povinny dodržovat pokyny k zacházení s léčivým přípravkem podle SPC. Přitom součástí SPC je vždy seznam terapeutických indikací, pro něž lze přípravek použít.
Výjimkou z této povinnosti dodržovat údaje obsažené v SPC, tedy i léčebné indikace, je použití přípravku podle § 8 odst. 3 až 5 zákona o léčivech. Zatímco ustanovení § 8 odst. 3 zákona o léčivech řeší podmínky předepisování a použití v ČR dosud neregistrovaných léčivých přípravků, ustanovení § 8 odst. 4 umožňuje použít přípravek již registrovaný způsobem off label za splnění stanovených podmínek. Podle tohoto ustanovení může ošetřující lékař, pokud není léčivý přípravek distribuován nebo není v oběhu léčivý přípravek potřebných terapeutických vlastností, použít registrovaný přípravek způsobem, který není v souladu se SPC (jde tedy o použití přípravku off label), je li však takový způsob dostatečně odůvodněn vědeckými poznatky.
Zákon o léčivech tedy umožňuje použití registrovaného přípravku způsobem off label pouze za splnění těchto podmínek:
- žádný léčivý přípravek potřebných terapeutických vlastností (který má v SPC danou terapeutickou indikaci) není registrován, nebo
- léčivý přípravek potřebných terapeutických vlastností sice je registrován, avšak není distribuován a současně
- použití přípravku off label je dostatečně odůvodněno vědeckými poznatky.
Zákon o léčivech dále stanoví (v § 8 odst. 5), že poskytovatel zdravotních služeb odpovídá podle právních předpisů za škodu na zdraví nebo za usmrcení člověka, k nimž došlo v důsledku použití neregistrovaného léčivého přípravku nebo použití registrovaného léčivého přípravku způsobem off label.
Typickým příkladem pro ukázku postupu podle ustanovení § 8 odst. 4 zákona o léčivech může být imatinib. Léčivý přípravek Glivec 100 mg má schválenou indikaci: „Glivec je indikován k léčbě dospělých a dětských pacientů s Ph+ CML v chronické fázi onemocnění, u kterých selhala léčba interferonem alfa, nebo kteří jsou v akcelerované fázi onemocnění nebo v blastické krizi,“ zatímco např. léčivý přípravek Imatinib Pharmagen 100 mg má schválenou a v SPC uvedenou indikaci: „Imatinib Pharmagen je indikován k léčbě dospělých pacientů s Ph+ CML v blastické krizi.“ Léčivý přípravek Imatinib Pharmagen tedy nemá v SPC uvedenu indikaci léčby dospělých pacientů s Ph+ CML v chronické fázi onemocnění, přičemž použití tohoto přípravku v dané indikaci nemůže byt v souladu s citovaným ustanovením § 8 odst. 4 zákona o léčivech; ačkoliv by takové použití bylo odůvodněno vědeckými poznatky, je registrován a současně je dostupný léčivý přípravek potřebných vlastností (tedy přípravek s touto indikací uvedenou v SPC).
Podobně lze identifikovat rozdíly v indikacích léčivého přípravku Xagrid („Xagrid je indikován ke snížení zvýšeného počtu trombocytů u rizikových pacientů s esenciální trombocytemií, u nichž došlo k intoleranci stávající léčby nebo tato léčba nesnížila zvýšený počet trombocytů na přijatelnou úroveň“) a léčivého přípravku Thromboreductin („Thromboreductin je indikován k léčbě esenciální trombocytemie u pacientů s vysokým rizikem, u nichž selhala nebo je neúčinná jiná cytoreduktivní terapie, či tuto terapii pacient netoleruje. Thromboreductin může být podán alternativně pacientům s vysokým rizikem i jako lék první volby“).
V této souvislosti je pro úplnost vhodné upozornit, že podle zákona o zdravotních službách (§ 28 odst. 1) je nutný svobodný a informovaný souhlas pacienta s tímto postupem. Navíc, občanský zákoník (zákon č. 89/2012 Sb., v platném znění) stanoví (v § 2950), že kdo se hlásí jako příslušník určitého stavu nebo povolání k odbornému výkonu nebo jinak vystupuje jako odborník, nahradí škodu, způsobí li ji neúplnou nebo nesprávnou informací nebo škodlivou radou danou za odměnu v záležitosti svého vědění nebo dovednosti.
Anagrelid – situace v ČR, registrace dvou léčivých přípravků s úplnou vlastní dokumentací
„Příběh“ anagrelidu započal roku 1973, kdy výzkumníci Beverung a Partyka patentovali (US3932407 A ze dne 19. listopadu 1973) syntézu látek ze skupiny quinazolin 2 onů, mezi něž anagrelid patří [3]. Patent na anagrelid byl poskytnut farmaceutické společnosti Bristol Myers. Příběh anagrelidu pak z hlediska patentu pokračoval až 30. července roku 1981. Tohoto dne byla přiřčena priorita (US4444777 A) výzkumníkům Flemingovi a Buyniskému [4], přičemž oba jmenovaní patent poskytli též společnosti Bristol Myers. Jednalo se o patent na formulaci lékové formy anagrelidu a sulfinpyrazonu. Zatímco sulfinpyrazon byl následně uveden na trh, v případě anagrelidu se tak nestalo.
Anagrelid byl, už jako trombocyty redukující léčivo, patentován a zaregistrován v zemích EU farmaceutickou společností AOP Orphan Pharmaceuticals AG pod názvem Thromboreductin. Farmaceutická společnost Shire zaregistrovala anagrelid pod názvem Agrylin ve Spojených státech amerických a následně pod názvem Xagrid jej registrovala EMA v zemích EU. Léčivému přípravku Agrylin byl v USA přiznán status tzv. orphan drug. Léčivý přípravek Thromboreductin byl v ČR registrován dne 30. dubna 2004 cestou národní registrace. Léčivý přípravek Xagrid byl v ČR (EU) registrován dne 16. listopadu 2004 cestou evropské registrace.
Přípravky Xagrid a Thromboreductin nejsou bioekvivalentní, a tudíž by neměly být v lékárnách zaměňovány. V roce 2009 totiž byla publikována (Petrides a kol.) [17] bioekvivalenční studie, která prokázala významně odlišnou biologickou dostupnost anagrelidu obsaženého v přípravcích Xagrid a Thromboreductin a z ní plynoucí rozdílnou bezpečnost. Zmíněná bioekvivalenční studie předznamenala další patent, jehož majitelem je společnost AOP Orphan Pharmaceuticals AG (EP2367539 ze dne 4. prosince 2009 [5]). Tento patent se váže k Thromboreductinu, přičemž patentované složení (včetně pomocných látek) vede k omezení maximálních plazmatických koncentrací, a tím i na dávce (na plazmatických koncentracích) závislých nežádoucích účinků anagrelidu.
Všechny výše uvedené patenty již exspirovaly. Výjimkou je posledně citovaný evropský patent EP2367539, který bude exspirovat až v prosinci roku 2029. V ČR budou v blízké budoucnosti nepochybně registrována generika anagrelidu ‒ všechna však podle názoru autorů patrně s odkazem na léčivý přípravek Xagrid, případně dle vlastní dokumentace či jinak než odkazem na Thromboreductin. V takovém případě žádné z nich s největší pravděpodobností nebude bioekvivalentní s Thromboreductinem.
Do zmíněné bioekvivalenční
studie (Petrides a kol.) [17] bylo zařazeno 24 zdravých
dobrovolníků (14 žen a 10 mužů) průměrného věku
23 ± 4 let, jimž byla ve dvojitě zaslepeném
a zkříženém uspořád![GRAF 1 Rozdíly v průběhu plazmatických koncentrací v čase mezi referenčním (Xagrid) a testovaným (Thromboreductin) přípravkem; volně podle [17] – Petrides, et al., 2009. cmax – maximální plazmatická koncentrace](https://www.remedia.cz/photo-a-31285---.jpg) ání (s odstupem 7 dnů)
vždy podána jednorázová dávka anagrelidu ve výši 2 mg
a k zapití byla podána voda v množství 240 ml.
Přípravky s obsahem anagrelidu byly vzájemně porovnávány
jako referenční léčivo (Xagrid) a jako testované léčivo
(Thromboreductin). Zjištěné rozdíly farmakokinetických
vlastností obou přípravků (plocha pod křivkou, maximální
plazmatické koncentrace ‒ cmax) byly statisticky vysoce
významné. Celková míra vstřebání anagrelidu obsaženého
v testovaném přípravku oproti referenčnímu přípravku byla
77 % (68‒86 % při 90% CI [interval spolehlivosti]; p < 0,001)
a hodnoty cmax anagrelidu obsaženého
v testovaném přípravku oproti referenčnímu přípravku byly
66 % (58‒76 % při 90% CI; p < 0,001).
Čas dosažení cmax (tmax) anagrelidu byl
v případě testovaného přípravku oproti referenčnímu
přípravku u o jednu hodinu prodloužen (graf 1). Obdobné změny byly pozorovány i u hlavního
metabolitu anagrelidu (3 hydroxyanagrelidu).
ání (s odstupem 7 dnů)
vždy podána jednorázová dávka anagrelidu ve výši 2 mg
a k zapití byla podána voda v množství 240 ml.
Přípravky s obsahem anagrelidu byly vzájemně porovnávány
jako referenční léčivo (Xagrid) a jako testované léčivo
(Thromboreductin). Zjištěné rozdíly farmakokinetických
vlastností obou přípravků (plocha pod křivkou, maximální
plazmatické koncentrace ‒ cmax) byly statisticky vysoce
významné. Celková míra vstřebání anagrelidu obsaženého
v testovaném přípravku oproti referenčnímu přípravku byla
77 % (68‒86 % při 90% CI [interval spolehlivosti]; p < 0,001)
a hodnoty cmax anagrelidu obsaženého
v testovaném přípravku oproti referenčnímu přípravku byly
66 % (58‒76 % při 90% CI; p < 0,001).
Čas dosažení cmax (tmax) anagrelidu byl
v případě testovaného přípravku oproti referenčnímu
přípravku u o jednu hodinu prodloužen (graf 1). Obdobné změny byly pozorovány i u hlavního
metabolitu anagrelidu (3 hydroxyanagrelidu).
U celkem 24 zdravých dobrovolníků
byl sledován výskyt nežádoucích účinků; ve zkříženém
uspořád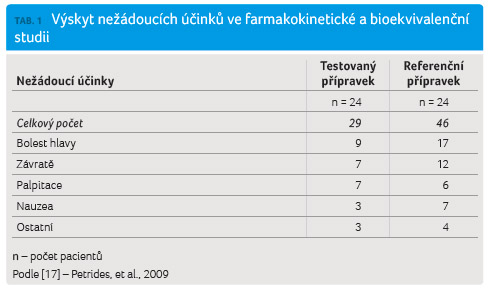 ání studie nejprve 24 zdravých dobrovolníků
dostalo referenční přípravek a následně 24 totožných
zdravých dobrovolníků testovaný přípravek. Celkový výskyt
nežádoucích účinků byl vyšší ve skupině s referenčním
přípravkem (46 nežádoucích účinků) než ve skupině
s testovaným přípravkem (29 nežádoucích účinků),
přičemž tento rozdíl byl statisticky významný (p < 0,05)
(tab. 1).
ání studie nejprve 24 zdravých dobrovolníků
dostalo referenční přípravek a následně 24 totožných
zdravých dobrovolníků testovaný přípravek. Celkový výskyt
nežádoucích účinků byl vyšší ve skupině s referenčním
přípravkem (46 nežádoucích účinků) než ve skupině
s testovaným přípravkem (29 nežádoucích účinků),
přičemž tento rozdíl byl statisticky významný (p < 0,05)
(tab. 1).
V této souvislosti je třeba uvést, že obdobných případů existuje více. Dobrým příkladem mohou být přípravky obsahující léčivou látku levotyroxin. V ČR je k dispozici více přípravků, z nichž Eltroxin a Euthyrox jsou registrovány na základě vlastní úplné dokumentace (tedy jako originály), Letrox je registrován na základě literárních údajů a Synthroxine je registrován tzv. hybridní registrací s odkazem na Euthyrox a s vlastní dokumentací. Ani jeden z uvedených léčivých přípravků nebyl zkoumán z hlediska bioekvivalence s dalšími. Nelze je tedy považovat za terapeuticky zaměnitelné. Obdobná situace je u levotyroxinů i v zahraničí. Nemůže proto překvapit, že analýzou 199 hlášení výskytu nežádoucích účinků v souvislosti s terapií levotyroxinem bylo v USA zjištěno [13], že ke 177 nežádoucím účinkům (tj. 88,9 %) došlo v důsledku substituce předepsaného levotyroxinu za jiný přípravek. Přitom pouze ve 13 případech byl lékař o substituci informován a ve 153 případech (tj. 86,4 %) informován nebyl. Celkem 54 nežádoucích účinků (ze zmíněných 199) bylo vyhodnoceno jako závažné a z nich 52 (tj. 96,3 %) se vyskytlo v souvislosti se substitucí.
Léčivé přípravky Xagrid a Thromboreductin tedy nejsou bioekvivalentní, proto nemohou být s Thromboreductinem bioekvivalentní ani generika odkazující se k registrační dokumentaci Xagridu. Na jednu stranu lze v budoucnu očekávat snahu regulačních orgánů tyto přípravky považovat za terapeuticky zaměnitelné z hlediska úhrady (tedy z hlediska zákona č. 48/1997 Sb., o veřejném zdravotním pojištění), zatímco z hlediska zákona č. 378/2007 Sb., o léčivech, tyto přípravky nemohou být považovány za zaměnitelné. V případě zmíněných přípravků dokonce existují relevantní důkazy, že nejen nejsou bioekvivalentní, ale že mají rovněž odlišnou snášenlivost plynoucí z rozdílného výskytu nežádoucích účinků, které jsou závislé na dávce (plazmatické koncentraci).
Porovnání výsledků kontrolovaných klinických studií s anagrelidem z hlediska účinnosti
Dosud nebylo provedeno žádné přímé srovnání účinnosti léčivých přípravků Xagrid a Thromboreductin formou randomizované, kontrolované a zaslepené klinické studie. Z velkého množství studií publikovaných s jednotlivými přípravky budou diskutována data, která by umožnila porovnání účinnosti. Takovým parametrem např. může být podávaná průměrná dávka.
Léčivé přípravky Xagrid/Agrylin
(v Evropě a v USA) a Thromboreductin byly
v minulosti předmětem řady publikací pojednávajících
o účinnosti a bezpečnosti terapie. Dosud nebyla
publikována žádná randomizovaná klinická studie u pacientů
s esenciální trombocytemií, v níž by byla porovnána
účinnost a bezpečnost obou zmíněných přípravků. Cílem
našeho srovnání není porovnání vlivu jednotlivých přípravků
na počet trombocytů, ale nepřímé porovnání průměrné
dávky použité v klinických studiích (tab. 2).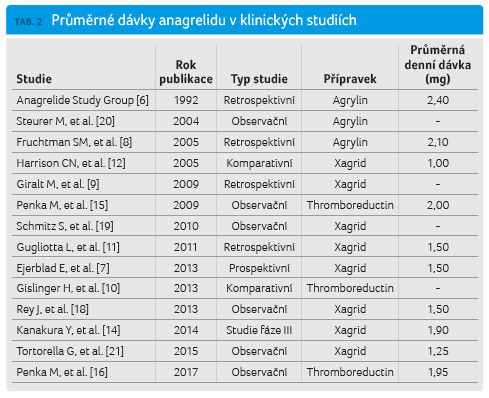
V celkem čtyřech studiích nebyly uvedeny průměrné dávky podávaného anagrelidu. Po přepočtu průměrné dávky připadající na Xagrid/Agrylin a Thromboreductin bylo zjištěno, že mezi těmito dávkami není žádný rozdíl a v případě obou přípravků dosahují hodnot 1,96 mg.
Porovnání výsledků kontrolovaných klinických studií s anagrelidem z hlediska bezpečnosti
Vzhledem k tomu, že z výsledků jediné přímé komparace léčivých přípravků Xagrid a Thromboreductin vyplývá odlišný výskyt nežádoucích účinků, budou z velkého množství studií publikovaných s jednotlivými přípravky diskutována data bezpečnosti, zejména výskyt nežádoucích účinků a podíl pacientů, u nichž bylo z důvodu jejich výskytu nutné terapii ukončit.
Přípravky Xagrid a Thromboreductin obsahují léčivou látku anagrelid, která vyvolává řadu nežádoucích účinků závislých na výši plazmatických koncentrací/dávky (například bolest hlavy, palpitace) a nezávislých na plazmatických koncentracích/dávce (např. otoky, parestezie). Výskyt nežádoucích účinků ‒ vedle účinnosti ‒ ovlivňuje dlouhodobou perzistenci k léčbě. U pacientů s dlouhodobým výskytem nežádoucích účinků dochází i přes dobrou terapeutickou účinnost ke snížení kvality života a terapie anagrelidem u nich může být z těchto důvodů ukončena. Snášenlivost anagrelidu je tedy klíčovým parametrem při dlouhodobé léčbě.
Klíčovou prací, která identifikovala rozdíly mezi Xagridem a Thromboreductinem, byla farmakokinetická studie publikovaná Petridesem a kol. (2009) [17]. Jak již bylo uvedeno výše, bylo v tomto sledování zaprvé prokázáno, že zmíněné léčivé přípravky nejsou bioekvivalentní (tj. mají odlišný průběh plazmatických koncentrací v čase), a zadruhé, že mají rozdílnou snášenlivost (tj. výskyt nežádoucích účinků závislých na plazmatických koncentracích/dávce).
Z důvodu případné identifikace
rozdílů v bezpečnosti jsme provedli analýzu výsledků 14
studií publikovaných v letech 1992‒2016. Výběr sledování
byl dán v nich obsaženými informacemi o výskytu
nežádoucích účinků. Podmínk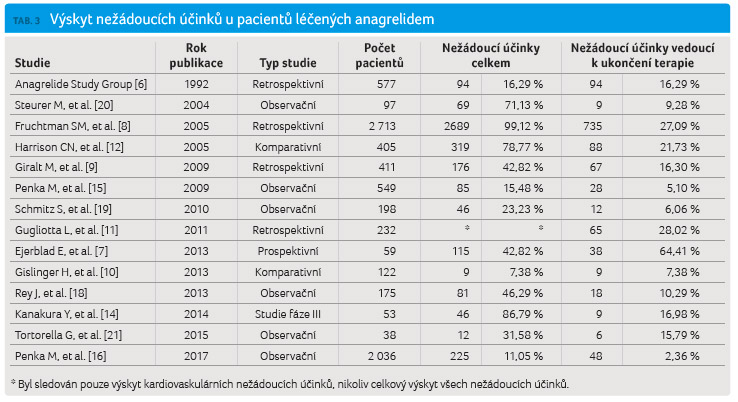 ou pro zařazení studie
do porovnání byla informace o podílu pacientů, kteří
ukončili terapii z důvodu výskytu nežádoucích účinků.
K porovnání byl použit též výstup informací z národního
registru, který ke dni 31. 12. 2016 obsahoval
informace o celkem 2 036 pacientech léčených anagrelidem.
Všech 14 hodnocených studií zahrnovalo celkem 7 665 pacientů.
U 4 509 z těchto nemocných (58,85 %) byl pozorován
nejméně jeden nežádoucí účinek. Z důvodu výskytu
nežádoucích účinků pak byla léčba u 1 122 nemocných
(14,64 %) ukončena.
ou pro zařazení studie
do porovnání byla informace o podílu pacientů, kteří
ukončili terapii z důvodu výskytu nežádoucích účinků.
K porovnání byl použit též výstup informací z národního
registru, který ke dni 31. 12. 2016 obsahoval
informace o celkem 2 036 pacientech léčených anagrelidem.
Všech 14 hodnocených studií zahrnovalo celkem 7 665 pacientů.
U 4 509 z těchto nemocných (58,85 %) byl pozorován
nejméně jeden nežádoucí účinek. Z důvodu výskytu
nežádoucích účinků pak byla léčba u 1 122 nemocných
(14,64 %) ukončena.
Ve většině studií uvedených v tabulce 3 byl podáván Xagrid (ve studiích provedených v USA Agrylin). Celkem 4 958 pacientů (64,68 %) bylo léčeno přípravkem Xagrid/Agrylin a 2 707 pacientů (35,31 %) bylo léčeno přípravkem Thromboreductin. Pokud byl porovnán výskyt nežádoucích účinků a podíl pacientů, u nichž byla terapie anagrelidem ukončena kvůli výskytu nežádoucích účinků uváděných v jednotlivých sledováních, zjišťujeme, že oba ukazatele byly statisticky významně vyšší při terapii Xagridem/Agrylinem (tab. 4, graf 2).
Rozd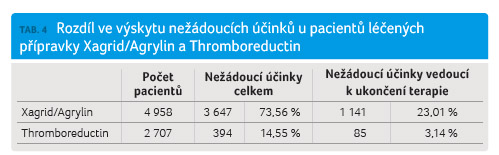 íly ve výskytu nežádoucích
účinků, ačkoliv jsou svými hodnotami výmluvné (14,55 %
v případě Thromboreductinu a 82,27 % v případě
Xagridu), nelze pro značný rozptyl údajů z jednotlivých
studií přeceňovat. Odlišná je situace při porovnání podílu
pacientů, kteří z důvodu výskytu nežádoucích účinků
terapii anagrelidem ukončili. Pouze u 3,14 % nemocných
léčených Thromboreductinem byla léčba ukončena z důvodu
výskytu nežádoucích účinků, zatímco z publikovaných
klinických a observačních studií s přípravkem
Xagrid/Agrylin to bylo 23
íly ve výskytu nežádoucích
účinků, ačkoliv jsou svými hodnotami výmluvné (14,55 %
v případě Thromboreductinu a 82,27 % v případě
Xagridu), nelze pro značný rozptyl údajů z jednotlivých
studií přeceňovat. Odlišná je situace při porovnání podílu
pacientů, kteří z důvodu výskytu nežádoucích účinků
terapii anagrelidem ukončili. Pouze u 3,14 % nemocných
léčených Thromboreductinem byla léčba ukončena z důvodu
výskytu nežádoucích účinků, zatímco z publikovaných
klinických a observačních studií s přípravkem
Xagrid/Agrylin to bylo 23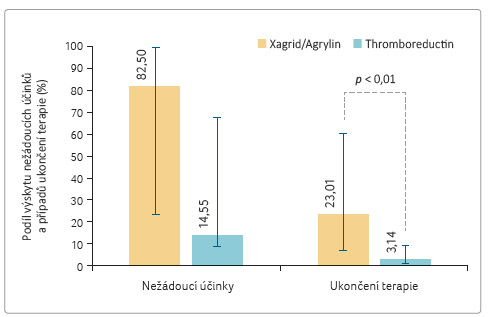 ,01 %. Tento rozdíl je statisticky
významný (p < 0,01).
,01 %. Tento rozdíl je statisticky
významný (p < 0,01).
Takové zjištění je v souladu s výše uvedenými informacemi publikovanými Petridesem a kol. [17]. V této práci bylo u 24 zdravých dobrovolníků pozorováno celkem 29 nežádoucích účinků při podání Thromboreductinu a celkem 46 nežádoucích účinků při podání Xagridu. Naše zjištění vyššího výskytu nežádoucích účinků a vyššího podílu pacientů, u nichž byla terapie pro nežádoucí účinky ukončena, je s tímto zjištěním plně konzistentní.
Léčivé přípravky Xagrid/Agrylin a Thromboreductin tedy nejsou terapeuticky zaměnitelné, protože nejsou bioekvivalentní, a současně mají odlišnou bezpečnost. Vyšší výskyt nežádoucích účinků je přitom spojen s častějším ukončením léčby z uvedených důvodů.
U budoucích generik anagrelidu je proto nezbytně nutné vědět, zda se jedná o generika léčivého přípravku Xagrid/Agrylin, nebo léčivého přípravku Thromboreductin, neboť u obou je silný důkaz svědčící o odlišné farmakokinetice a snášenlivosti.
Vzhledem ke skutečnosti, že podle názoru autorů je Thromboreductin příštích několik let chráněn patentem, je vysoce pravděpodobné, že generika anagrelidu, která se v budoucnu nepochybně objeví na tuzemském trhu, budou (pravděpodobně) registrována odkazem na registrační dokumentaci Xagridu. Při záměně léčivých přípravků Xagrid (a jeho generik) a Thromboreductin (a jeho generik) je tedy nezbytně nutné mít na paměti, že může dojít ke změně snášenlivosti léčby a že oba přípravky nelze považovat za terapeuticky totožné.
Zjišťování, zda je konkrétní léčivý přípravek generikem, nebo zda byl registrován jinou registrační procedurou, může být v podmínkách ČR vskutku detektivní prací, zatímco Štátny ústav pre kontrolu liečiv v Bratislavě (www.sukl.sk) takové informace zveřejňuje ‒ formou Odkazovaná žiadosť ‒ liek rovnocenný s referenčným liekom (generická) v případě generik a formou Samostatná úplná – nové liečivo v případě přípravků registrovaných na základě vlastní úplné registrační dokumentace u originálních léčiv. Státní ústav pro kontrolu léčiv v Praze (www.sukl.cz) takové informace bohužel neuvádí.
Shrnutí, závěr a doporučení
Častý argument prezentovaný ekonomickým managementem zdravotnických zařízení, že na prvním místě jsou úspory, nemůže obstát. Prvořadým cílem je zajistit konkrétnímu pacientovi optimální lék a současně přitom zajistit lékaři dostatečné bezpečí, že neporušil některou z řady povinností, jež mu rozsáhlá legislativa ukládá (např. postup off label při nesplnění některé z podmínek § 8 odst. 4 zákona o léčivech atd.).
Preskribující lékař by při výběru léčivého přípravku před jeho předpisem neměl pouze spoléhat na skutečnost, že obsahuje jistou konkrétní léčivou látku. Měl by se snažit zjistit (je li to ovšem objektivně v jeho možnostech), zda zamýšlený léčivý přípravek je u konkrétního pacienta vhodný z hlediska zaměnitelnosti.
Zaměnitelnost lze chápat ve dvojím smyslu ‒ zaměnitelnost daná právním rámcem a zaměnitelnost daná vlastnostmi léčivé látky v konkrétním přípravku.
Při zaměnitelnosti dané právním rámcem je zcela nezbytné, aby lékař předepsal přípravek, který má v platném SPC uvedenou shodnou indikaci (pro niž lékař zamýšlí léčivý přípravek předepsat). V opačném případě lékař může postupovat off label se všemi hmotněprávními důsledky uvedenými výše. Příkladem jsou např. odlišná znění indikací uvedených v SPC přípravků Thromboreductin a Xagrid.
Při zaměnitelnosti dané vlastnostmi léčivé látky v konkrétním přípravku se vychází z principů bioekvivalence a za zaměnitelná se považují taková léčiva, která právě mají pozitivní průkaz bioekvivalence. V případě, že takový průkaz neexistuje, nebo dokonce existuje pozitivní průkaz non bioekvivalence, nemělo by vůbec k „volné“ záměně docházet a změna terapie musí být spojena s častějším a pečlivějším monitoringem pacienta. V lékárně při výdeji léčivých přípravků, které nejsou bioekvivalentní, by ke generické substituci nikdy nemělo dojít bez předchozího výslovného souhlasu lékaře.
Seznam použité literatury
- [1] Thromboreductin. AOP Orphan, datum registrace: 19. 6. 2015. Souhrn údajů o přípravku. Státní ústav pro kontrolu léčiv. Dostupné na: http://www.sukl.cz/modules/medication/download.php?file=SPC80525.pdf&type=spc&as=thromboreductin‑spc
- [2] Xagrid. Shire Pharmaceutical Contracts Ltd, datum registrace: 18. 6. 2015. Souhrn údajů o přípravku. Státní ústav pro kontrolu léčiv. Dostupné na: http://www.ema.europa.eu/docs/cs_CZ/document_library/EPAR_‑_Product_Information/human/000480/WC500056557.pdf
- [3] US3932407. Dostupné na: http://www.google.com/
- [4] US4444777. Dostupné na: http://www.google.ch/
- [5] EP2367539. Dostupné na: http://patentimages.storage.googleapis.com/pdfs/3f53117ccceea7d5833d
- [6] Anagrelide Study Group. Anagrelide, a therapy for thrombocythemic states: experience in 577 patients. Am J Med 1992; 92: 69–76.
- [7] Ejerblad E, Kvasnicka HM, Thiele J, et al. Diagnosis according to World Health Organization determines the long‑term prognosis in patients with myeloproliferative neoplasms treated with anagrelide: results of a prospective long‑term follow‑up. Hematology 2013; 18: 8–13.
- [8] Fruchtman SM, Petitt RM, Gilbert HS, et al.; Anagrelide Study Group. Anagrelide: analysis of long‑term efficacy, safety and leukemogenic potential in myeloproliferative disorders. Leuk Res 2005; 29: 481–491.
- [9] Giralt M, Navas V, Hernández‑Nieto L, et al.; en representación del Grupo de Estudio de Enfermedades Mieloproliferativas Filadelfia Negativas (GEMFIN). Retrospective analysis of the efficacy and tolerability of anagrelide in patients with essential thrombocytemia: Spanish registry of essential thrombocytemia. Med Clin (Barc) 2009; 133: 86–90.
- [10] Gisslinger H, Gotic M, Holowiecki J, et al.; ANAHYDRET Study Group. Anagrelide compared with hydroxyurea in WHO‑classified essential thrombocythemia: the ANAHYDRET Study, a randomized controlled trial. Blood 2013; 121: 1720–1728.
- [11] Gugliotta L, Tieghi A, Tortorella G, et al. Low impact of cardiovascular adverse events on anagrelide treatment discontinuation in a cohort of 232 patients with essential thrombocythemia. Leuk Res 2011; 35: 1557–1563.
- [12] Harrison CN, Campbell PJ, Buck G, et al.; United Kingdom Medical Research Council Primary Thrombocythemia 1 Study: Hydroxyurea compared with anagrelide in high‑risk essential thrombocythemia. N Engl J Med 2005; 353: 33–45.
- [13] Hennessey JV, Malabanan AO, Haugen BR, Levy EG. Adverse event reporting in patients treated with levothyroxine: results of the pharmacovigilance task force survey of the american thyroid association, american association of clinical endocrinologists, and the endocrine society. Endocr Pract 2010; 16: 357–370.
- [14] Kanakura Y, Miyakawa Y, Wilde P, et al. Phase III, single‑arm study investigating the efficacy, safety, and tolerability of anagrelide as a second‑line treatment in high‑risk Japanese patients with essential thrombocythemia. Int J Hematol 2014; 100: 353–360.
- [15] Penka M, Schwarz J, Ovesná P, et al.; CZEMP. Essential thrombocythaemia and other myeloproliferative disorders with thrombocythaemia treated with Thromboreductin. A report from the database of register for the 1st quarter of 2010. Vnitr Lek 2010; 56: 503–512.
- [16] Penka M, Schwarz J, Ovesná P; IBU MU Brno. Léčba esenciální trombocytémie a dalších myeloproliferativních chorob anagrelidem. Registr českých pacientů léčených Thromboreductinem. CZEMP symposium, 9. 6. 2017, Praha (údaje z registru ke dni 31. 12. 2016).
- [17] Petrides PE, Gisslinger H, Steurer M, et al. Pharmacokinetics, bioequivalence, tolerability, and effects on platelet counts of two formulations of anagrelide in healthy volunteers and patients with thrombocythemia associated with chronic myeloproliferation. Clin Ther 2009; 31: 386–398.
- [18] Rey J, Viallard JF, Keddad K, et al.; FOX study investigators. Characterization of different regimens for initiating anagrelide in patients with essential thrombocythemia who are intolerant or refractory to their current cytoreductive therapy: results from the multicenter FOX study of 177 patients in France. Eur J Haematol 2014; 92: 127–136.
- [19] Schmitz S, Stauch M, Schlag R. Anagrelide for the treatment of thrombocythaemia in daily clinical practice: a post‑marketing observational survey on efficacy and safety performed in Germany. Onkologie 2010; 33: 39–44.
- [20] Steurer M, Gastl G, Jedrzejczak WW, et al. Anagrelide for thrombocytosis in myeloproliferative disorders: a prospective study to assess efficacy and adverse event profile. Cancer 2004; 101: 2239–2246.
- [21] Tortorella G, Piccin A, Tieghi A, et al.; Gimema Foundation project “Registro Italiano Trombocitemie (RIT)”. Anagrelide treatment and cardiovascular monitoring in essential thrombocythemia. A prospective observational study. Leuk Res 2015; 39: 592–598.