Transtyretinová srdeční amyloidóza a možnosti její léčby
Souhrn:
Hradec J. Transtyretinová srdeční amyloidóza a možnosti její léčby. Remedia 2020; 30: 86–90.
Transtyretin je protein, syntetizovaný především v játrech, který normálně tvoří tetramery. V důsledku genových mutací nebo s přibývajícím věkem se mohou molekuly transtyretinu špatně poskládat (ztratit svoji normální prostorovou konformaci) a ukládat se v srdci a jiných orgánech jako amyloid. Srdeční postižení se při transtyretinové amyloidóze (ATTR) typicky manifestuje pseudohypertrofií levé srdeční komory a/nebo srdečním selháním se zachovanou ejekční frakcí. ATTR kardiomyopatie je relativně vzácné onemocnění, které má špatnou prognózu. Dosud neexistovala žádná účinná specifická farmakologická léčba, pouze léčba symptomatická, většinou diuretiky pro úlevu od dušnosti. Léčebné možnosti nyní zahrnují několik nových léků, které zasahují do amyloidogeneze na různých úrovních a různými mechanismy. Na základě pozitivních výsledků klinické studie ATTR ACT byl nedávno pro léčbu ATTR kardiomyopatie registrován lék tafamidis, který stabilizuje transtyretinové tetramery. V pokročilé fázi klinického výzkumu jsou také léky patisiran a inotersen, které inhibují syntézu transtyretinu.
Summary:
Hradec J. Transthyretin cardiac amyloidosis and possibilities of its treatment. Remedia 2020; 30: 86–90.
Transthyretin is a protein synthetized mostly by the liver, which normaly forms tetramers. As a result of gene mutations or as an ageing related phenomenon, transthyretin molecules may disfold (loose their normal space conformation) and deposit in the heart or in other organs as amyloid. Cardiac involvement in transthyretin related cardiomyopathy manifests typically as left ventricular pseudohypertrophy and/or heart failure with preserved ejection fraction. ATTR cardiomyopathy is relatively rare disease with bad prognosis. Untill recently no effective specific pharmacological tratment did exist, just symptomatic treatment, mostly by diuretics for dyspnoe relief. The therapeutic options now include several new drugs targeting amyloidogenesis at different steps and by different mechanisms. Following the positive results of ATTR ACT study, tafamidis was recently registered for the treatment of ATTR cardiopmyopathy. This drug stabilizes the transthyretin tetramers. There are also transthyretin synthesis inhibiting drugs patisiran and inotersen in an advanced phase of clinical research.
Key words: amyloid cardiomyopathy, transthyretin amyloidosis, clinical study ATTR ACT, tafamidis, patisiran, inotersen
Úvod
Amyloidóza je onemocnění, při němž dochází k ukládání
špatně nebo chybně konformovaných proteinů do extracelulárního prostoru
různých tkání a orgánů. To narušuje jejich strukturu a funkci.
Amyloid je špatně složený (angl. misfolded) protein, jehož sekundární
struktura, tedy lokální prostorové uspořádání polypeptidových řetězců, je
narušeno. Nejčastěji se jedná o vznik tzv. beta‑struktury skládaného
listu. Takto špatně složené proteiny mají silnou tendenci tvořit špatně
rozpustné nebo zcela nerozpustné agregáty s buď fibrilárním, nebo
hrudkovitým (amorfním) charakterem a ty se jako amyloid ukládají
do mezibuněčného prostoru. Proto se amyloidóza také označuje jako
konformační onemocnění nebo také anglickým termínem „protein misfolding disease“.
Nejedná se tedy o klinicko‑patologickou jednotku, ale o obecný
patologický fenomén spojující heterogenní skupinu onemocnění s různými
příčinami, různou symptomatologií, odlišnou prognózou a také
s odlišnou léčbou, jejichž společnou vlastností je ukládání amyloidu [1]. V současnosti je
identifikováno asi 30 lidských bílkovin, které se mohou stát prekurzorem
amyloidu (mají amyloidogenní potenciál).
Amyloidózy se dělí podle různých klinických hledisek
na primární (idiopatické) nebo sekundární (reaktivní nebo zánětlivé),
na hereditární nebo získané, na systémové nebo orgánové
(lokalizované). Dnes je jednoznačně preferována klasifikace biochemická podle
amyloidogenního proteinu. Nejznámější a zřejmě také nejčastější je AL‑amyloidóza,
způsobená ukládáním lehkých řetězců imunoglobulinů, buď jako primární
amyloidóza při monoklonální gamapatii nejistého významu, nebo jako sekundární
AL‑amyloidóza při mnohočetném myelomu nebo maligním lymfomu. Podobně vzniká AH‑amyloidóza
jako systémové onemocnění při ukládání těžkých řetězců imunoglobulinů nebo AA‑amyloidóza
z ukládání sérového amyloidu A při chronických zánětlivých
onemocněních, jako je např. Crohnova nemoc nebo revmatoidní artritida [2].
Ukládání amyloidu v myokardu může vést ke vzniku
kardiomyopatie. U nás představuje amyloidóza nejčastější formu
restriktivní kardiomyopatie.
Transtyretinová amyloidóza (ATTR)
Transtyretinová amyloidóza je nejčastější formou hereditární
amyloidózy (ATTRv – variantní), obvykle je systémová. Příčinou je mutovaný
transtyretin (TTR). Transtyretin je transportní protein pro tyroxin
a retinol, který je primárně tvořen v játrech. Při elektroforetickém
dělení plazmatických bílkovin se TTR pohybuje před albuminem, a proto se
také označuje jako prealbumin. Termíny ATTR a prealbuminová amyloidóza
jsou synonyma. Transtyretinový gen je lokalizován na chromozomu 18
a je kódován čtyřmi exony. Mutace se mohou objevit v exonech 2, 3
a 4. Dosud je popsáno kolem 120 mutací, které mají za následek ATTR
s velmi variabilní klinickou prezentací, od čisté polyneuropatie
s autonomní dysfunkcí (familiární autonomní polyneuropatie, FAP) přes
smíšené neurologické a kardiologické postižení až k selektivnímu
postižení srdce pod obrazem amyloidové kardiomyopatie. Většinou jde
o substituci jednoho nukleotidu. Nejčastější jsou substituce Val30Met,
Val122Ile a Val94Ala. S ATTR kardiomyopatií je nápadně často spojena
mutace Val122Ile, která se nachází u 3,4 %
Afroameričanů. Mimo USA je nejčastější mutací Val30Met s fenotypem, který
se zřetelně regionálně mění. Endemicky se tato varianta vyskytuje
v Portugalsku, Švédsku, Japonsku a v některých oblastech Jižní
Ameriky [3].
Za normálních podmínek se transtyretin vyskytuje
v séru jako tetramer složený ze čtyř identických monomerů. Při
pozměněné konformaci podléhá obtížně proteolýze a vytváří nerozpustná
amyloidová depozita.
Hereditární amyloidózy včetně ATTR jsou autozomálně
dominantní onemocnění s různým stupněm penetrance a obvykle
s pomalou progresí. Většinou jde o heterozygotní stavy, homozygotní
stavy mají těžší a závažnější průběh [1].
Prevalence ATTR se odhaduje na 1 : 100 000, ale výskyt regionálně
značně kolísá. Je vyšší např. u Afroameričanů, v severním Portugalsku
a v severní Africe. Je také pravděpodobné, že s cíleným
screeningem, např. u lidí s echokardiograficky prokázanou hypertrofií
levé komory, bude odhalováno stále více nemocných s ATTR. Příznaky se
u hereditární ATTR začnou zpravidla objevovat až ve středním věku.
Maximální koncentrace amyloidu je u hereditární formy v myokardu
a v periferních nervech. Zda dojde k poškození srdce
a eventuálnímu rozvoji ATTR kardiomyopatie, záleží na typu mutace.
Varianta s mutací Met30Val, která je u nás nejčastější, často
způsobuje poruchy srdečního rytmu, dysfunkci sinusového uzlu, blokády
Tawarových ramének či úplnou síňokomorovou blokádu, která si obvykle vyžádá
trvalou kardiostimulaci. Infiltrativní postižení myokardu vede k rozvoji
ATTR kardiomyopatie, která je echokardiograficky nerozlišitelná
od postižení myokardu při AL‑amyloidóze. Manifestuje se srdečním selháním
se zachovanou ejekční frakcí. Prognóza symptomatických nemocných s ATTR
kardiomyopatií je obecně špatná. Udává se však, že je přece jen o něco
lepší než prognóza nemocných s AL‑amyloidózou srdce. Je to způsobeno
rozdíly v histologické formě ukládání amyloidu v srdci a dalšími,
dosud neobjasněnými příčinami [2].
Na vzniku ATTR se může podílet také tzv. divoký typ
(wild‑type) transtyretinu, který představuje přirozenou formu proteinu
a vyskytuje se v každém lidském organismu a obvykle nepůsobí
žádný patologický stav. Tento typ amyloidózy je považován za získanou
formu (ATTRwt) a nazývá se senilní amyloidóza. Senilní ATTR postihuje
zejména srdce, ale i plíce, cévní stěnu a karpální tunel. Vyskytuje
se u starších osob, typicky ve věku nad 80 let, častěji u mužů.
Symptomatologie ATTR bývá charakteristická pro každou
specifickou mutaci. V klinickém obraze obvykle dominuje periferní
senzomotorická a autonomní polyneuropatie. Postižení ledvin je mnohem méně
časté, někdy mohou dominovat příznaky postižení gastrointestinálního traktu (zejména
průjmy a úbytek tělesné hmotnosti), vyvolávající příčinou bývá obvykle
dysfunkce autonomních nervů.
U koho by se mělo po srdeční ATTR aktivně pátrat?
Rozhodně u nemocných se symptomy srdečního selhání nejasné etiologie, se
synkopami v anamnéze nebo s bradyarytmiemi, kteří mají současně
echokardiografický obraz hypertrofie levé srdeční komory nejasné etiologie
a/nebo srdeční selhání se zachovanou ejekční frakcí. Základem diagnostiky je
echokardiografické vyšetření. Diagnózu pak potvrdí speciální scintigrafické
vyšetření s 99mTc‑značenými deriváty fosfátů nebo
nejpřesněji průkaz transtyretinových amyloidových depozit
z endomyokardiální biopsie [4].
Léčba ATTR kardiomyopatie
Donedávna žádná specifická léčba ATTR kardiomyopatie
neexistovala. Nemocní s městnavým srdečním selháním byli léčeni pouze
symptomaticky, především diuretiky a eventuálně blokátory
mineralokortikoidních receptorů. Průlom v léčbě přišel v roce 2018,
kdy byly publikovány výsledky klinické studie ATTR‑ACT s tafamidisem [5]. První skutečně účinný
lék tafamidis však již není jedinou možností léčby ATTR kardiomyopatie, ale
postupně se objevují další zajímavé léčebné postupy. Rozdělme je podle
mechanismu účinku.
Stabilizace tetramerů transtyretinu
Tato stabilizace probíhá prostřednictvím farmakologických
stabilizátorů, tzv. chaperonů (z franc. gardedáma).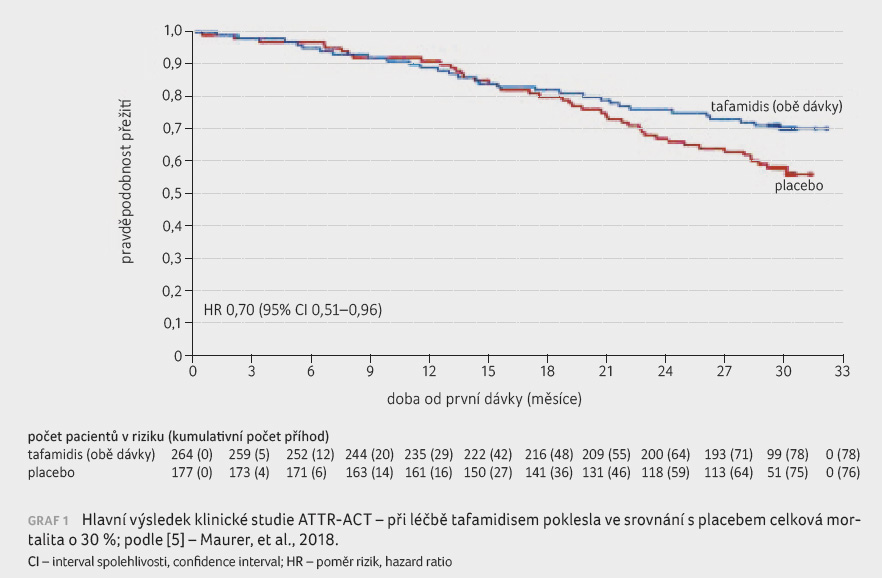 Chaperony jsou
farmaka, která napomáhají správnému poskládání proteinů a udržují tak
jejich náležitou sekundární strukturu (konformaci). Prvním z takovýchto
specifických stabilizátorů transtyretinu je tafamidis.
Tafamidis je chemicky benzoxazol (obr. 1), malá
molekula, která inhibuje disociaci transtyretinových tetramerů vazbou
na vazebná místa tyroxinu. Je to první chorobu modifikující lék schválený
pro léčbu ATTR (Vydaqel®, Pfizer), nejprve ve Spojených státech
amerických v roce 2011 a v Japonsku v roce 2013 pro léčbu
FAP. Později, po pozitivním výsledku klinické studie ATTR‑ACT, přiznal
americký Úřad pro kontrolu potravin a léčiv (Food and Drug Administration,
FDA) tafamidisu status průlomové léčby a v roce 2019 ho jako „orphan
drug“ registroval také pro léčbu ATTR kardiomyopatie. Ve stejném roce byl
registrován pro léčbu ATTR kardiomyopatie rovněž v Evropské unii.
Chaperony jsou
farmaka, která napomáhají správnému poskládání proteinů a udržují tak
jejich náležitou sekundární strukturu (konformaci). Prvním z takovýchto
specifických stabilizátorů transtyretinu je tafamidis.
Tafamidis je chemicky benzoxazol (obr. 1), malá
molekula, která inhibuje disociaci transtyretinových tetramerů vazbou
na vazebná místa tyroxinu. Je to první chorobu modifikující lék schválený
pro léčbu ATTR (Vydaqel®, Pfizer), nejprve ve Spojených státech
amerických v roce 2011 a v Japonsku v roce 2013 pro léčbu
FAP. Později, po pozitivním výsledku klinické studie ATTR‑ACT, přiznal
americký Úřad pro kontrolu potravin a léčiv (Food and Drug Administration,
FDA) tafamidisu status průlomové léčby a v roce 2019 ho jako „orphan
drug“ registroval také pro léčbu ATTR kardiomyopatie. Ve stejném roce byl
registrován pro léčbu ATTR kardiomyopatie rovněž v Evropské unii.
V klinické studii ATTR‑ACT bylo randomizováno celkem
441 nemocných s transtyretinovou kardiomyopatií k užívání dvou
dávek tafamidisu (20 a 80 mg)
nebo placeba po dobu 30 měsíců. Ve srovnání s placebem vedly obě
dávky tafamidisu k poklesu celkové mortality o 30 % (poměr rizik [HR] 0,70; 95% interval spolehlivosti,
confidence interval; [CI] 0,51–0,96) a k významně nižšímu počtu
kardiovaskulárních hospitalizací (HR 0,68; 95% CI 0,56‒0,81), graf 1. Léčba
tafamidisem vedla rovněž ke zmírnění symptomů, což se projevilo zlepšením
tolerance zátěže při šestiminutovém testu chůzí a zlepšením kvality života
hodnocené dotazníkem KCCQ (Kansas City Cardiomyopathy Questionnaire; pro obojí p < 0,001)
[5].
Lék byl také dobře tolerován se srovnatelným výskytem
nežádoucích účinků.
Většímu rozšíření této nadějné léčby však brání neuvěřitelně
vysoká cena tafamidisu. Katalogová cena roční léčby je v USA 225 000 dolarů, což z něj činí nejdražší
kardiovaskulární lék. V Evropě, konkrétně v Portugalsku, je sice cena
roční léčby „jen“ třetinová, ale stále velmi vysoká – 60 000 dolarů [6].
Další selektivní stabilizátor (chaperon) transtyretinu byl
označen kódovým označením AG10. Lék byl zkoušen
u 49 nemocných se symptomatickou ATTR kardiomyopatií ve fázi II
klinického zkoušení. Pozitivní výsledky vedly k naplánování klinické
studie fáze III, která měla být zahájena v roce 2019 [3].
V roce 2006 bylo oznámeno, že nesteroidní
antirevmatikum diflunizal se váže na vazebné místo
transtyretinu, stabilizuje jeho tetramery a in vitro
brání tvorbě amyloidových fibril. Zdá se však, že klinický výzkum v tomto
směru nepokračuje [3].
Inhibice syntézy mutovaného transtyretinu
Velmi účinnou léčbou hereditární ATTR je ortotopická
transplantace jater. U získané formy však účinná není.
U mladších nemocných s ATTR kardiomyopatií přichází v úvahu
rovněž kombinovaná transplantace jater a srdce. Z pochopitelných
důvodů se však tato léčba může týkat jen ojedinělých nemocných [3].
Další možností je farmakologická inhibice
exprese genu pro transtyretin. V klinickém zkoušení je hned několik
látek.
Patisiran je malá interferující RNA, která blokuje
expresi transtyretinu jak variantního, tak i divokého typu. Látka je
enkapsulována v lipidových nanočásticích, které cílí do jater,
a podává se v intravenózní infuzi. V klinické studii fáze III
APOLLO, která zahrnula 225 nemocných s ATTR polyneuropatií, vedl patisiran
k významnému zlepšení neurologického stavu [7]. Příznivé výsledky této
studie vedly jak v USA, tak i v EU k registraci patisiranu
(Onpattro®,
Alnylam) pro léčbu dospělých nemocných s ATTR polyneuropatií. Nemocní
ve studii APOLLO s echokardiograficky doloženým postižením srdce
(n = 126) měli prokazatelné zlepšení echokardiografických parametrů
a pokles plazmatické koncentrace NT‑proBNP (N‑terminálního natriuretického
propeptidu typu B). To dokládá, že patisiran zpomaluje zhoršování funkce
levé srdeční komory a navozuje její příznivou remodelaci [7].
Jinou malou signální RNA je revusiran,
který byl testován na 200 nemocných s ATTR kardiomyopatií
v klinické studii fáze III ENDEAVOUR. Studie hodnotila toleranci zátěže
pomocí šestiminutového testu chůzí. Byla ukončena předčasně pro nárůst
mortality [3].
Antisense
oligonukleotid inotersen inhibuje tvorbu obou
forem transtyretinu, jak variantní (v), tak divokého typu (wt).
V multicentrické randomizované klinické studii fáze III NEURO‑TTR byl
zkoušen inotersen u 172 nemocných s ATTR polyneuropatií. Lék
zpomalil progresi neurologického postižení a byl dobře tolerován [8]. Na základě těchto
výsledků schválila FDA inotersen pod obchodním názvem Tegsedi®
(Akcea) pro léčbu ATTR polyneuropatie. Ve studii NEURO‑TTR mělo 63 % nemocných také
kardiomyopatii, podskupinová analýza však neměla dostatečnou statistickou sílu
na to, aby prokázala příznivý vliv na postižení srdce [8].
Inhibice agregace oligomerů a jejich disrupce
Epigalokatechin‑3‑galát (EGCG) je
nejrozšířenějším katechinem v zeleném čaji. Váže se na rozpustný
transtyretin a snižuje tím pravděpodobnost disociace tetramerů; inhibuje tak agregaci
oligomerů do amyloidových fibril a podporuje jejich disagregaci.
V malé klinické studii u 35 nemocných se srdeční ATTR nedošlo
po 12 měsících léčby ani ke zlepšení echokardiografických parametrů,
ani k poklesu plazmatických koncentrací NT‑proBNP [3].
Degradace a reabsorpce amyloidových fibril
Několik hydrofobních molekul je účinných
v destabilizaci a dezintegraci amyloidových fibril, což usnadňuje
reabsorpci uloženého amyloidu makrofágy. Takové vlastnosti mají in vitro např. tetracyklinová antibiotika včetně doxycyklinu. Kombinace doxycyklinu s kyselinou
tauroursodeoxycholovou (TUDCA) má ještě silnější účinek, který vede
ve zvířecích modelech dokonce k úplnému vyplavení amyloidu
z tkání. Výsledky malé klinické studie fáze II u 28 nemocných
s ATTR však nebyly přesvědčivé [3].
Dalším možným léčebným postupem jsou protilátky
proti sérovému amyloidu P (SAP). SAP je normální plazmatický
glykoprotein, který stabilizuje a chrání amyloidové fibrily před
proteolytickou degradací. Miridesap je protilátka
vážící se na cirkulující SAP a navozující tak jeho hepatální
clearance. Tato protilátka byla testována v malé studii, která však byla
v roce 2018 ukončena a miridesap již není dále klinicky rozvíjen [3].
Nově však byl zahájen klinický výzkum monoklonální
protilátky specificky zaměřené na depozita ATTR, jejíž kódové označení je PRX004 [3].
Další terapeutické možnosti
I přes nadějný rozvoj specifické léčby ATTR
kardiomyopatie zůstává stále základem léčba symptomatická včetně podpůrných
nefarmakologických postupů. Z farmakoterapie se jedná o léky podávané
podle evropských i českých doporučení pro léčbu srdečního selhání [9,10]. Protože se
v naprosté většině případů ATTR kardiomyopatie jedná o srdeční
selhání se zachovanou ejekční frakcí, je farmakoterapie vesměs symptomatická.
Betablokátory jsou často špatně
tolerovány či pro hypotenzi a/nebo převodní poruchy úplně kontraindikovány.
Podobně mohou být pro hypotenzi špatně tolerovány i inhibitory systému renin‑angiotenzin‑aldosteron (RAAS). Diuretika jsou při dušnosti a plicní kongesci
naprosto nezbytná. Často a bezpečně jsou rovněž používány blokátory mineralokortikoidních receptorů. Nemocní se srdeční
amyloidózou mají vyšší tromboembolické riziko, dokonce i při sinusovém
rytmu. Je proto u nich vždy třeba zvážit antikoagulační
léčbu. Infiltrace myokardu amyloidem predisponuje ke vzniku
bradyarytmií a převodních poruch. U vybraných nemocných je proto
indikována trvalá kardiostimulace. Pacienti mají též
vyšší riziko náhlé srdeční smrti, většinou pro maligní tachyarytmie.
V indikovaných případech je namístě implantace
kardioverteru‑defibrilátoru (ICD), ať již v rámci sekundární, nebo
primární prevence. Je třeba počítat s vyššími defibrilačními prahy. Implantace levokomorových mechanických podpor má vysokou
krátkodobou mortalitu a horší výsledky než při dilatační kardiomyopatii.
Izolovaná transplantace srdce byla u nemocných
s ATTR kardiomyopatií provedena jen ojediněle.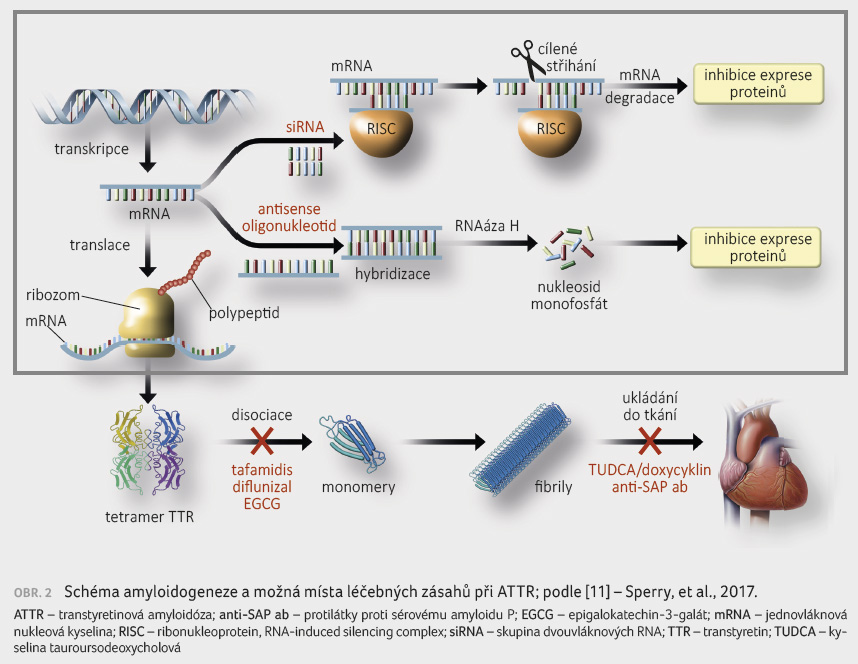
Shrnutí
Závěrem lze konstatovat, že zatímco donedávna neexistovala
pro nemocné se srdeční ATTR žádná tzv. evidence based léčba, situace se začíná
dramaticky měnit. Pozitivní výsledky vedly k registraci prvního
specifického léku pro léčbu ATTR kardiomyopatie tafamidisu. Hlubší porozumění
patogenezi ATTR pak vedlo k navržení a klinickému zkoušení dalších
léčebných strategií cílených na jiné kroky amyloidogenní kaskády (obr. 2) [11].
Nejnadějnějšími léky se zdají být patisiran a inotersen, jež
blokují syntézu transtyretinu a jeví se ještě účinnějšími než tafamidis,
který stabilizuje transtyretinové tetramery. Zdá se, že cestou
do budoucnosti bude kombinace léků s komplementárními mechanismy účinku.
Tomu ale prozatím brání příliš vysoká cena. Péče o nemocné se srdeční ATTR
proto nepochybně bude muset být soustředěna do několika vysoce
specializovaných center.
Seznam použité literatury
- [1] Gomesová B. Amyloidózy – molekulární a buněčné mechanismy, laboratorní diagnostika. Bakalářská práce. Přírodovědecká fakulta, Masarykova univerzita, Brno, 2004.
- [2] Adam Z, Krejčí M, Simonides J. Choroby způsobené ukládáním monoklonálních imunoglobulinů. Postgraduální medicína 2011; 13: 1009‒1017.
- [3] Emdin M, Aimo A, Rapezzi S, et al. Treatment of cardiac transthyretin amyloidosis: An update. Eur Heart J 2019; 40: 3699‒3706.
- [4] Paleček T, Krejčí J. Studie ATTR ACT. Přelom v léčbě transtyretinové srdeční amyloidózy. Interv Akut Kardiol 2019; 18: 45‒48.
- [5] Maurer MS, Schwartz JH, Gundapaneni B, et al., for the ATTR ACT Study Investigators. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med 2018; 379: 1007‒1016.
- [6] Wending P. Pfizerʼs $225K price for heart drug tafamidis slammed. Medscape, Jan 15, 2020.
- [7] Adams D, Gonzales Duarte A, O’Riordan WD, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med 2018; 379: 11‒21.
- [8] Benson MD, Waddington Cruz M, Berk JL, et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med 2018; 379: 22‒31.
- [9] Ponikowski P, Voors AA, Anker S, et al. 2016 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure. The Task Force for the diagnosis and treat-ment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J 2016; 37: 1‒17.
- [10] Špinar J, Hradec J, Špinarová L, Vítovec J. Souhrn Doporučených postupů ESC pro diagnostiku a léčbu akutního a chronického srdečního selhání z roku 2016. Připraven Českou kardiologickou společností. Cor Vasa 2016; 58: 597‒636.
- [11] Sperry BW, Tang WWH. Amyloid heart disease: genetics translated into disease modifying therapy. Heart 2017; 103: 812‒817.