Trombotická trombocytopenická purpura
Souhrn:
Trombotická trombocytopenická purpura je vzácné, klinicky závažné onemocnění ze skupiny trombotických mikroangiopatií s vysokou mortalitou bez včasné adekvátní léčby. Je charakterizována pentádou příznaků (trombocytopenie, mikroangiopatická hemolytická anémie, neurologické příznaky, horečka a renální selhání), která však ve většině případů není kompletně vyjádřena. Příčinou onemocnění je těžký deficit depolymerázy von Willebrandova faktoru ADAMTS13. Laboratorní stanovení deficitu je součástí diagnostických kritérií. Ve většině případů se jedná o získaný deficit vznikající především v dospělosti a vyvolaný vznikem autoprotilátek proti enzymu. Vzácný vrozený deficit je způsoben mutacemi v genu pro ADAMTS13. Diferenciálně diagnosticky je třeba odlišit především hemolyticko‑uremický syndrom, některé syndromy spojené s těhotenstvím, diseminovanou intravaskulární koagulaci a trombotickou mikroangiopatii v rámci některých systémových onemocnění. Léčbou první volby v případě získané trombotické trombocytopenické purpury jsou výměnné plazmaferézy, souběžně je podávána imunosupresivní léčba kortikosteroidy. Po dosažení remise je nutná dlouhodobá dispenzarizace pacientů pro trvalé riziko relapsu onemocnění.
Key words: thrombotic thrombocytopenic purpura – thrombotic microangiopathy – ADAMTS13 deficiency – plasma exchange.
Summmary:
Thrombotic thrombocytopenic purpura (TTP) is a rare, aggressive form of thrombotic microangiopathy. It has a high mortality unless timely appropriate therapy is initiated. It’s classically characterized with a pentad of symptoms (thrombocytopenia, microangiopathic hemolytic anemia, neurologic abnormalities, fever and renal failure). However, the majority of patients do not have all five clinical features. The underlying pathological mechanism that causes TTP is a severely decreased activity of von Willebrand factor depolymerase, ADAMTS13. The laboratory demonstration of profound decrease in enzyme activity confirms the diagnosis of TTP. The deficiency is acquired in most cases and is a result of an autoantibody against ADAMTS13. A rare congenital deficiency is caused by mutations in ADAMTS13 gene. A careful evaluation of the patient is necessary in order to distinguish TTP from other thrombotic microangiopathy syndromes, notably hemolytic‑uremic syndrome, some pregnancy‑related disorders, disseminated intravascular coagulation and thrombotic microangiopathy associated with some systemic disorders. The first line treatment in acquired TTP is plasma exchange. Immunosuppresive treatment with corticorsteroids is administered concomitantly. After a remission occurs, patients need to be followed up in long term for the risk of subsequent relapse.
Úvod
Trombotická trombocytopenická purpura (TTP) je závažná forma trombotické mikroangiopatie, která vede k multiorgánové dysfunkci způsobené trombotizací a následnou ischemií v mikrocirkulaci. Jedná se o vzácné onemocnění (incidence se dle různých pramenů pohybuje mezi 1–6 případy/milion obyvatel/rok) [1]. Jeho klinická závažnost spočívá zejména ve vysokém riziku úmrtí – mortalita se před zavedením účinné léčby pohybovala kolem 90 % [2]. Naopak, při správné a včas zahájené léčbě lze u téměř 90 % pacientů dosáhnout remise onemocnění.
První případ byl popsán už v roce 1924 (syndrom Moschowitzové). Enzym ADAMTS13, který je v patofyziologii onemocnění klíčový a jehož stanovení je dnes součástí diagnostických kritérií, byl objeven až v roce 1998 (Furlan, Tsai). V posledních letech probíhá intenzivní výzkum na molekulárně genetické úrovni, díky němuž se prohlubuje úroveň poznání patofyziologického mechanismu a výrazně se přibližuje možnost cílené terapie onemocnění.
Patofyziologie
Onemocnění TTP je charakterizováno tvorbou hyalinních trombů v terminálních arteriolách a kapilárách, jejíž příčinou je těžký deficit metaloproteinázy ADAMTS13 (a disintegrin and metalloprotease with thrombospondin type 1 repeats, synonymum: depolymeráza von Willebrandova faktoru). Fyziologická role tohoto enzymu spočívá ve štěpení vysoce hemostaticky aktivních ultravelkých multimerů von Willebrandova faktoru (UL vWF). Von Willebrandův faktor (vWF) je jednou z centrálních molekul hemostázy. V případě ruptury cévní stěny mj. slouží jako most mezi subendoteliální tkání a trombocyty, které pak vytvářejí primární trombus. Von Willebrandův faktor je uvolňován z endotelií ve formě UL vWF multimerů, které jsou následně štěpeny enzymem ADAMTS13 za podmínek vysokého smykového tření, typického pro arterioly a kapiláry. Při hlubokém deficitu ADAMTS13 se multimery UL vWF kumulují a nekontrolovaně na sebe váží krevní destičky, což vede ke vzniku trombů v mikrocirkulaci a k následné orgánové ischemii. Dochází ke spotřebování trombocytů a k rozvoji střední až těžké trombocytopenie. Destičkové tromby v malých cévách vytvářejí síť, o niž se mechanicky rozbíjejí erytrocyty, což vede k hemolytické anémii se zvýšeným počtem úlomků erytrocytů – schistocytů.
Získaná TTP
Deficit ADAMTS13 je ve většině
případů (zhruba 95 %) způsoben přítomností protilátky,
která enzym váže a blokuje jeho aktivitu – vzniká tak
získaná forma TTP. Jedná se většinou o protilátku třídy
IgG namířenou proti regionu, který je zásadní pro funkci
proteázy. Onemocnění je častější u žen. Medián věku
stanovení diagnózy se v různých souborech pohybuje kolem 40
let, s velkým věkovým rozptylem (např. Oklahoma Registry
uvádí 9–78 let) [3]. Získaná TTP může být buď
primární, nebo – vzácněji – může vzniknout
sekundárně v rámci jiného systémového onemocnění
(autoimunitní onemocnění, nádory, infekce) či v souvislosti
s expozicí některým lékům (např. tiklopidin, chinin,
cyklosporin). Část případů je diagnostikována v souvislosti
s těhotenstvím (zpravidla ve třetím trimestru) či
peripartálně; v těchto případech je nutné odlišit TTP
od preeklampsie a HELLP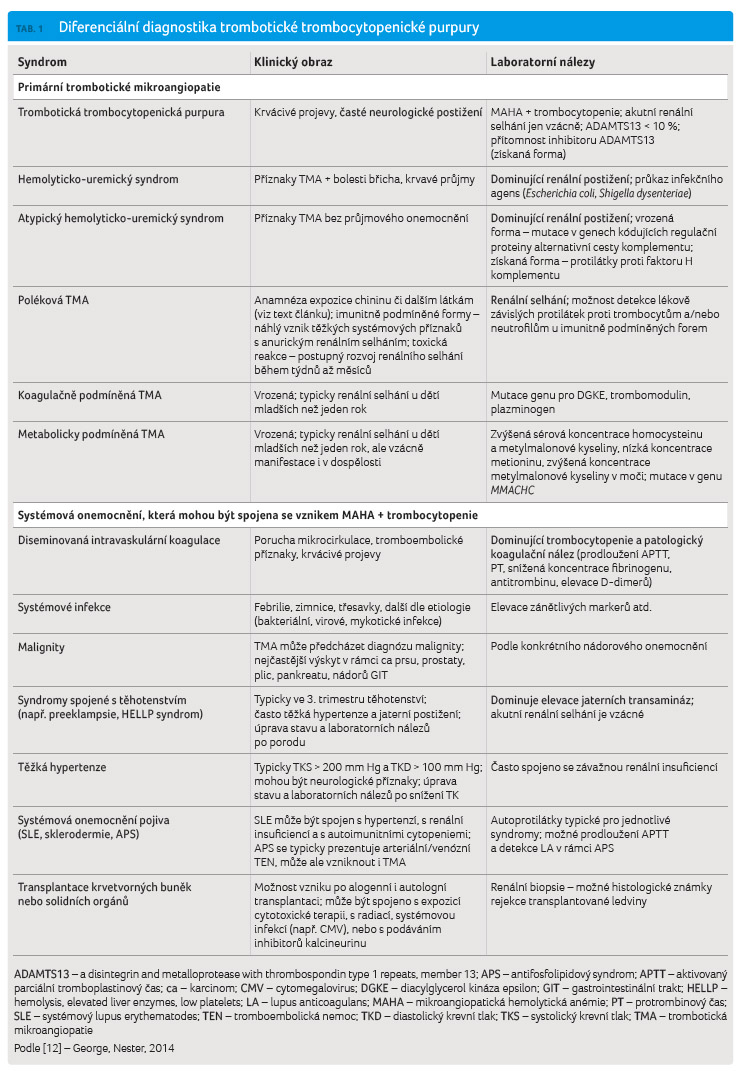 syndromu (viz tab. 1).
syndromu (viz tab. 1).
Vrozená TTP
Vrozený deficit ADAMTS13 (synonyma – kongenitální TTP, hereditární TTP, familiární TTP, Upshawův–Schulmanův syndrom) je velmi vzácné onemocnění (tvoří zhruba 5 % případů diagnostikované TTP) s autozomálně recesivní dědičností. Je způsobené mutacemi v genu pro ADAMTS13. Jedná se o pestrou škálu mutací (záměnné mutace, nonsense mutace, inzerce, delece) na různých místech genu. Vyskytují se buď v dvojitě heterozygotním, či v homozygotním stavu [4]. Genetické variabilitě kongenitální TTP odpovídá rozmanitost klinického obrazu, ať už se jedná o tíži onemocnění, či věk prvních projevů. Více než polovina pacientů je diagnostikována v dětství (většina z nich v novorozeneckém věku), u části pacientů se však první příznaky onemocnění objeví až v dospělosti. Mezi pacientkami, u nichž je onemocnění diagnostikováno v souvislosti s těhotenstvím, mohou případy vrozené TTP tvořit až 25 % [5].
Stanovení diagnózy
Klinický obraz, laboratorní nálezy
Onemocnění TTP je klasicky charakterizováno pentádou příznaků: trombocytopenie, mikroangiopatická hemolytická anémie, neurologické příznaky, horečka a renální selhání.
Trombocytopenie je důsledkem konzumpce destiček v mikrovaskulárních trombech. Jedná se o středně těžkou až těžkou trombocytopenii (medián počtu trombocytů v různých studiích je 10–30 × 109/l), která se může manifestovat kožními hematomy, petechiemi, menoragií, epistaxí, hematurií či krvácením z gastrointestinálního traktu. Závažné až život ohrožující krvácení je spíše vzácné.
Postižení mikrocirkulace centrální
nervové soustavy se prezentuje často bolestmi hlavy či
zmateností, může se objevit přechodná ložisková léze,
epileptiformní příznaky, ale i iktus či kóma. Renální
insuficience, je li přítomna, je spíše mírná, akutní
renální selhání je vzácné. U pacientů s TTP se dále
mohou objevit gastrointestinální příznaky (nespecifické bolesti
břicha, nauzea, zvracení, průjem) nebo kardiální postižení
(od bolestí na hrudi přes arytmie po akutní projevy
ischemie myokardu). Koagulační nález je u pacientů s TTP
zpravidla normální, mohou být přítomny nespecifické známky
aktivace koagulace (mírné prodloužení základních koagulačních
časů, mírná elevace D dimerů). Jak ukázala analýza dat
z různých registrů, uvedená kompletní klasická pentáda
příznaků je přítomna pouze u malého procenta pacientů.
Zatímco trombocytopenie a hemolytická anémie jsou nalézány
vždy, neurologické příznaky má 60–80 % pacientů,
porucha renálních funkcí je zjišťována v necelé polovině
případů a horečka je přítomna zhruba u jedné
čtvrtiny pacientů (tab. 2).
Vzhledem k tomu, že pro přežití pacientů s TTP je
klíčová včasná terapie a stanovení aktivity ADAMTS13
zpravidla není dostupné ihned, je dostatečnou indikací k zahájení
léčby přítomnost trombocytopenie a mikroangiopatické
hemolytické anémie zároveň s absencí jiných příčin
trombotické mikroangiopatie (viz odstavec Diferenciální
diagnostika).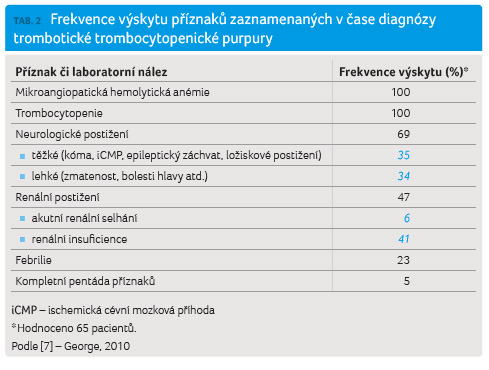
Stanovení aktivity inhibitoru ADAMTS13
Aktivita ADAMTS13 je stanovována z plazmy nebo séra pacienta. Principem většiny komerčně dostupných metod je fluorescenční detekce štěpení části vWF, která obsahuje štěpné místo pro depolymerázu ADAMTS13.
Stanovení inhibitoru ADAMTS13 se ve většině laboratoří provádí na stejném principu jako stanovení inhibitoru koagulačního faktoru VIII u pacientů s hemofilií – tzv. směsným testem. Plazma pacienta je smíchána s normální plazmou, a je li přítomen inhibitor ADAMTS13, blokuje aktivitu enzymu v normální plazmě. Pomocí série ředění plazmy pacienta, při nichž ještě probíhá inhibice depolymerázy, je pak určen titr inhibitoru.
Diagnózu TTP potvrzuje nález hlubokého deficitu ADAMTS13 (pod 10 %). V případě, že zároveň není přítomen inhibitor ADAMTS13, jedná se o vrozenou formu TTP. Aby byl výsledek vyšetření ADAMTS13 spolehlivý, je třeba provést odběr vzorku krve k vyšetření před zahájením léčby, jejíž součástí je podávání plazmy.
Diferenciální diagnostika
Trombotická mikroangiopatie je histologická diagnóza, která se klinicky a laboratorně manifestuje kombinací trombocytopenie + mikroangiopatické hemolytické anémie. Kromě TTP je typickým nálezem u dalších primárních onemocnění, ale vyskytuje se i v řadě systémových onemocnění. Aktivita ADAMTS13 může být v řadě těchto stavů sekundárně snížená, ale zůstává vyšší než 10 % [6]. V praxi však toto vyšetření vzhledem k jeho omezené dostupnosti spíše zpětně potvrdí diagnózu, která musí být iniciálně určena na základě kontextu rozvoje onemocnění, klinického obrazu a základních laboratorních vyšetření. Diferenciální diagnostika těchto často závažných stavů může být obtížná, je však zásadní pro správnou volbu terapeutického přístupu k pacientům.
Mezi primární onemocnění, která je třeba odlišit od TTP, patří především hemolyticko uremický syndrom (HUS), kde zpravidla dominuje renální postižení. Vyskytuje se buď v rámci infekce Shigella dysenteriae nebo infekce některými sérotypy Escherichia coli (shiga toxin HUS), nebo jde o onemocnění způsobené hereditárním či získaným deficitem regulačních proteinů alternativní cesty komplementu, např. tzv. faktoru H (tzv. atypický HUS).
Poléková trombotická mikroangiopatie (drug induced thrombotic microangiopathy) byla poprvé popsána v souvislosti s expozicí chininu. Mohou ji způsobit i např. některá protinádorová chemoterapeutika (gemcitabin, oxaliplatina, mitomycin), imunosupresiva (cyklosporin, takrolimus), inhibitory vaskulárního endoteliálního růstového faktoru (bevacizumab, sirolimus) či zneužívané látky (kokain).
Vznik trombotické mikroangiopatie v kontextu systémových onemocnění či syndromů byl zatím nejlépe popsán u následujících: komplikace těhotenství (preeklampsie, HELLP syndrom), maligní hypertenze, některé závažné infekce (např. HIV, cytomegalovirus), malignity. Nález mikroangiopatické hemolytické anémie plus trombocytopenie je i součástí diseminované intravaskulární koagulace, kde však dominuje patologický koagulační nález.
Diferenciální diagnostika TTP je přehledně shrnuta v tabulce 1.
Terapie
Získaná TTP je jedním z mála urgentních stavů v hematologii. Vzhledem k vysoké závažnosti onemocnění z hlediska morbidity a mortality a vzhledem k často rychlému rozvoji klinických příznaků je zahájení léčby nutné do 24 hodin od stanovení diagnózy TTP či při důvodném podezření na tuto diagnózu na základě klinického obrazu a výsledků základních laboratorních vyšetření (viz výše). Léčba by měla iniciálně probíhat ve specializovaných centrech s možností akutního zajištění výměnných plazmaferéz, v podmínkách jednotek intenzivní péče.
Výměnné plazmaferézy (TPE, total plasma exchange) jsou v dnešní době léčbou první volby získané TTP. Podle různých pramenů je doporučována výměna 1,0–1,5 plazmatického objemu denně [1,7]. Co se týče různých typů plazmy používané k hrazení volumu v rámci TPE, nebyly zjištěny rozdíly v bezpečnosti a účinnosti mezi jednotlivými plazmatickými produkty (K plazma, čerstvě zmražená plazma, solvent detergent ošetřená plazma) [7]. Zavedení plazmaferéz do léčby TTP znamenalo výrazné snížení rizika úmrtí pacientů. Provádění plazmaferéz je výrazně účinnější ve srovnání s podáváním samotné plazmy, zřejmě proto, že plazmaferéza umožňuje jednak odstranit z plazmy pacienta protilátky blokující aktivitu ADAMTS13, jednak pomáhá doplnit chybějící enzym dodáním volumu dárcovskou plazmou [8]. Samotná aplikace plazmy (v dávce 30–40 ml/kg) představuje dnes v rámci iniciální léčby pacienta pouze přechodnou léčebnou eventualitu do doby, kdy je možné zajistit provádění plazmaferéz. Výměnné plazmaferézy je nutné provádět denně do normalizace počtu trombocytů, vymizení známek aktivní hemolýzy a ústupu neurologických příznaků. Anémie se upravuje většinou pomaleji, ke zlepšení renálních funkcí, jsou li postiženy, dochází nejpozději [7].
V rámci terapie získané formy TTP s přítomností inhibitoru ADAMTS13 je souběžně s TPE zahajována imunosupresivní léčba kortikosteroidy, například metylprednisolonem 5–10 mg/kg denně podávaným po 3–5 dnů s následným snižováním dávek.
V případě refrakterity pacienta na iniciální léčbu lze zvýšit frekvenci plazmaferéz na 2× denně či potencovat imunosupresivní léčbu podáním rituximabu (375 mg/m2 1× týdně po dobu 4–8 týdnů) či vinkristinu (2 mg i.v. týdně, 4–6 dávek) [7–9].
U pacientů s vrozenou formou TTP jsou akutní epizody léčeny samotným podáváním plazmy v iniciální dávce 10–15 ml/kg [4]. Imunosupresivní léčba není indikována vzhledem k nepřítomnosti inhibitoru ADAMTS13. U některých pacientů je nutné dlouhodobé preventivní podávání plazmy v intervalu 2–4 týdny k doplnění funkčního enzymu. V současné době je ve fázi klinického zkoušení rekombinantní humánní ADAMTS13, který by měl být primárně určen pro pacienty s kongenitální TTP.
Prognóza
Po dosažení remise TTP pacienti zůstávají nadále v dispenzarizaci hematologického centra vzhledem k celoživotnímu riziku relapsu onemocnění. Frekvence relapsu se pohybuje v různých souborech mezi 20–50 %. Relaps je v současné době definován jako epizoda akutní TTP po více než 30 dnech remise onemocnění. Měření hodnot ADAMTS13 u pacientů v remisi ukázalo, že u pacientů s normální aktivitou ADAMTS13 je relaps nepravděpodobný, nicméně u části pacientů je pozorována dlouhodobě nízká aktivita enzymu pod 10 % bez klinických a laboratorních známek onemocnění [10].
Kromě rizika relapsu onemocnění jsou pacienti s TTP zřejmě dlouhodobě ohroženi zvýšenou morbiditou a mortalitou ve srovnání s ostatní populací, jak ukázala analýza dat z Oklahoma Registry. Bylo zjištěno mírné zhoršení kognitivních funkcí (snížená manuální zručnost, schopnost soustředění, jazyková výbavnost), zvýšená prevalence hypertenze, závažné deprese, systémového lupus erythematodes a albuminurie [11].
Závěr
Trombotická trombocytopenická purpura zůstává i v dnešní době závažnou diagnózou s mortalitou, která je bez včasné a dostatečně intenzivní léčby vysoká. Po dosažení remise onemocnění je prognóza pacientů zatížena vysokým rizikem relapsu. Pacienti by měli být léčeni a sledováni ve specializovaných centrech s dostupností plazmaferéz a se zkušenostmi s péčí o tyto pacienty.
Seznam použité literatury
- [1] Scully M, Hunt BJ, Benjamin S, et al. Guidelines on the diagnosis and management of thrombotic thrombocytopenic purpura and other thrombotic microangiopathies. Br J Haematol 2012; 158: 323–335.
- [2] Ridolfi RK, Bell RW. Thrombotic thrombocytopenic purpura: report of 25 cases and review of the literature. Medicine 1981; 60: 413–428.
- [3] George JN, Cuker A. Acquired TTP: Clinical manifestations and diagnosis. UpToDate, 2017, dostupné na www.uptodate.com.
- [4] George JN, Cuker A. Hereditary thrombotic thrombocytopenic purpura. UpToDate, 2017, dostupné na www.uptodate.com.
- [5] Moatti‑Cohen M, Garec C, Wolf M. et al. Unexpected frequency of Upshaw/Schulman syndrome in pregnancy‑onset of thrombotic thrombocytopenic purpura. Blood 2012; 119: 5888.
- [6] George JN, Nester C. Approach to the patient with suspected TTP, HUS or other thrombotic microangiopathy (TMA). UpToDate, 2017. Dostupné na: www.uptodate.com.
- [7] George JN. How I treat patients with thrombotic thrombocytopenic purpura: 2010. Blood 2010; 116: 4060–4069.
- [8] Blombery P, Scully M. Management of thrombotic thrombocytopenic purpura: current perspectives. J Blood Med 2014; 5: 15–23.
- [9] Salaj P. Poruchy hemostázy. Praha: Maxdorf Jessenius, 2017; 38–41.
- [10] Page EE, Kremer Hovinga JA, Terrell DR, et al. Clinical importance of ADAMTS13 activity during remission in patients with acquired thrombotic thrombocytopenic purpura. Blood 2016; 128: 2175–2178.
- [11] Vesely SK. Life after acquired thrombotic thrombocytopenic purpura: morbidity, mortality, and risks during pregnancy. J Thromb Haemost 2015; 13 (Suppl. 1): S216–S222.
- [12] George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Eng J Med 2014; 371: 654–666.