Zpráva z kongresu American Academy of Neurology 2014 – zaměřeno na roztroušenou sklerózu
Článek předkládá výsledky vybraných prací prezentovaných na 66. kongresu Americké neurologické akademie, který se konal na přelomu dubna a května 2014 ve Filadelfii v USA. Zejména jsou představeny novinky týkající se léčby roztroušené sklerózy.
Úvod
„We hold these truths to be self-evident, that all men are created equal, …“, úvodní citát z Deklarace nezávislosti vítal na letišti účastníky 66. ročníku Americké neurologické akademie (AAN), která se letos konala ve dnech 26. dubna až 3. května v kolébce americké demokracie ve Filadelfii v Pensylvánii.Kongresu se účastnili lékaři a výzkumníci z celého světa, vlastní jednání bylo rozděleno do řady sekcí, z nichž podstatná část byla věnována právě autoimunitním onemocněním, zejména roztroušené skleróze (RS).Během kongresu bohužel není možné navštívit všechna jednání a prohlédnout všechny postery, článek tedy rozhodně nemá ambici podat komplexní přehled všech nových informací z AAN, spíše jde o shrnutí informací ze zajímavých přednášek a posterů, které jsme měly možnost shlédnout.
Dominujícími tématy v sekci demyelinizačních onemocnění byly výsledky lékových studií, a to jak léků již registrovaných, tak nových léčiv, která jsou ve stadiu klinického zkoušení. Důraz byl přitom kladen nejen na účinnost jednotlivých přípravků, ale současně i na jejich bezpečnost. Dále byly prezentovány novinky o etiopatogenezi RS, zejména vliv environmentálních faktorů, které se podílejí na vzniku nemoci a jsou potenciálně preventabilní.
Léky první volby
Glatiramer acetát
Glatiramer acetát (GA) je kopolymer čtyř aminokyselin, které jsou součástí myelinového bazického proteinu. V současnosti registrovaná dávka 20 mg 1krát denně podkožně (GA20) je užívána od své registrace v roce 1995. Dávka 40 mg 3krát týdně podkožně (GA40) je registrována v USA od počátku roku 2014.
Dr. Jerry Wolinsky z Texasu prezentoval výsledky studie GLACIER (GLatiramer Acetate low frequenCy safety and patIent ExpeRience), která porovnávala bezpečnost a snášenlivost dvou zmíněných odlišných dávek přípravku. Do studie GLACIER bylo zařazeno 210 pacientů s relaps-remitentní RS (RR RS) celkem v 31 centrech v USA, kteří byli léčeni aslepoň šest měsíců dávkou GA20 1krát denně podkožně a následně byli randomizováni pro podávání dávek G20 nebo GA40 a sledováni v měsíci 1, 2 a 4.
Výsledky studie jsou povzbudivé. Prokázaly, že vyšší dávka s nižší frekvencí podávání vedla až k 50% redukci nežádoucích účinků vázaných přímo na aplikaci podkožní injekce a k redukci lokálních reakcí v místě vpichu. Snížení frekvence aplikace injekcí a tím potenciální redukce reakcí v místě vpichu je samozřejmě velmi významné, protože u řady pacientů je právě lokální kožní reakce příčinou ukončení léčby.Dále byla prezentována data z otevřené pokračovací fáze studie GALA (Glatiramer Acetate Low-frequency Administration). Do původní studie bylo zařazeno 1404 pacientů s RR RS, kteří byli náhodně randomizováni v poměru 1 : 1 do skupiny GA40 nebo do skupiny podávání placeba. Po 12 měsících došlo v léčené skupině k 34% redukci počtu relapsů ve srovnání s placebem (průměrná roční míra relapsů v léčené větvi byla 0,331 vs. 0,505 v placebové větvi; p < 0,0001). Současně došlo k významné redukci počtu gadolinium vychytávajících lézí a nově se zvětšujících lézí v T2 váženém obraze na magnetické rezonanci – MR (p < 0,0001). Dávka GA40 3krát týdně byla bezpečná a dobře tolerovaná.
Výsledky 24měsíční otevřené pokračovací fáze studie GALA prezentoval Dr. Omar Khan z Detroitu. Skupina pacientů léčených od počátku GA40 ve srovnání s původně placebovou skupinou, která byla převedena na léčbu GA40 až po 12 měsících, měla signifikantně lepší výsledky ve všech sledovaných parametrech – průměrná roční míra relapsů (0,18 vs. 0,26; risk ratio, poměr rizik – RR: 0,69; p = 0,0002), počet pacientů bez relapsu a bez šestiměsíční potvrzené progrese. Vedlejší nežádoucí účinky dávky GA40 byly obecně mírné. Závěrem lze shrnout, že dávka GA 40 mg 3krát týdně podkožně má účinnost srovnatelnou s původní dávkou GA 20 mg 1krát denně podkožně, ale tolerance léčby touto dávkou je lepší. Předpokládaná registrace GA40 v Evropě je na přelomu let 2014–2015.
Pegylovaný interferon beta-1a
Staronovým přípravkem, který bude zřejmě v blízkém časovém horizontu uveden do klinické praxe, je pegylovaný interferon beta-1a (pegIFN β-1a). Jde o upravenou molekulu původního intereferonu beta-1a (podávaného jednou týdně nitrosvalově). Tato nová úprava umožnuje podání podkožní a v delším intervalu. Pegylovaný interferon byl testován ve studii ADVANCE, což byla multicentrická, mezinárodní dvojitě slepá studie s účastí 1516 pacientů, kteří byli náhodně randomizováni v poměru 1 : 1 : 1 do jedné ze tří skupin: pegIFN β-1a 125 μg subkutánně každé dva týdny (2QW) nebo každé čtyři týdny (4QW) nebo placebo. Po prvním roce studie byli pacienti z placebové větve randomizováni do jedné ze dvou skupin léčených pegylovaným interferonem, pacienti dostávající původní léčbu v ní pokračovali beze změny i ve druhém roce.
Během prvního roku studie došlo u obou skupin pacientů léčených pegIFN β-1a k významnému zpomalení vývoje 12týdenní potvrzené progrese invalidity, a to o 38 % oproti placebu. Současně došlo ke snížení počtu relapsů za rok o 36 % vůči placebu ve skupině 2QW a o 28 % ve skupině 4QW. Dr. Peter Calabresi z Baltimoru prezentoval dvouletá klinická a bezpečnostní data. Celkově lze říci, že pacienti léčení po celou dobu dvou let měli lepší výsledky ve srovnání s pacienty, kteří v prvním roce dostávali placebo.
Dále skupina s vyšší frekvencí podávání léku (2QW) dosáhla lepších výsledků než při frekvenci podávání 4QW. Obě dávkovací schémata byla dobře tolerována. Důležitým výsledkem je nízká imunogenicita pegIFN β-1a.
Neutralizační protilátky byly detekovány u méně než 1 % pacientů, což je v souladu s předpokladem, že pegylace snižuje imunogenicitu přípravku. Klinický efekt léku byl podpořen i výsledky měření pomocí MR, jež prezentoval Dr. Douglas Arnold z Montrealu. Obě dávkovací schémata prokázala lepší výsledek ve srovnání s placebem při hodnocení kombinovaného parametru „clinical and MRI freedom from measured disease activity (FMDA)“, což je kombinované hodnocení výskytu klinické a MR aktivity.
Registrace léku se předpokládá na přelomu let 2014–2015, a to pravděpodobně v dávce 125 μg subkutánně 1krát za 2 týdny. Nicméně vzhledem k velmi dlouhým jednáním o úhradě v České republice lze reálně předpokládat dostupnost léku pro české pacienty až koncem roku 2015.
Perorální přípravky
Fingolimod
Perorálně podávaný fingolimod (0,5 mg denně) je v České republice k dispozici od října 2012 a je indikován (obdobně jako lék natalizumab) jako druhá volba léčby pacientů s RR RS v případě selhání léků první volby, případně jako lék první volby při agresivním průběhu RR RS s vysokým rizikem rychlého nárůstu invalidity. Výsledky studií týkající se tohoto přípravku prezentované na letošním AAN byly zaměřeny mimo jiné na prodloužené fáze základních klinických studií fáze III (FREEDOMS a FREEDOMS II, které zkoumaly účinek fingolimodu v dávce 0,5 mg a 1,25 mg podávané jednou denně oproti placebu, a studie TRANSFORM, kde byl podáván fingolimod oproti komparátoru interferonu β-1a
aplikovanému 1krát týdně nitrosvalově). Pacienti byli sledování až 60 měsíců od vstupního zařazení do studie fáze III.
Měření ročního procentuálního úbytku mozkové hmoty na MR ukázalo přetrvávající pozitivní trend i při takto prodlouženém intervalu sledování, byť byla studie limitována nestejným dávkováním léku u jednotlivých skupin pacientů ve výše uvedených studiích. Dalším parametrem, který byl zpracován v post hoc analýze v intervalu sledování 24, 36 a 48 měsíců u pacientů ze stejných tří základních studií fáze III s fingolimodem, byla invalidita měřená na stupnici EDSS (Expanded Disability Status Scale). Při zahrnutí všech pacientů léčených pouze dávkou fingolimodu 0,5 mg denně (zpracováno Kaplanovou-Meierovou křivkou) se dosáhlo lepšího či stejného skóre EDSS ve srovnání se vstupní hodnotou při zařazení do studie: 67,9 % (po 24 měsících sledování), 64,7 % (po 36 měsících sledování) a 66,8 % (po 48 měsících sledování).
Pozitivní výsledky z reálné praxe v podobě studie PANGAEA (Post-Authorization Non-interventional German sAfety study of GilEnyA in RRMS patients) představil kolektiv německých autorů Dr. Tjalfa Ziemssena z Drážďan. V souladu s indikačními kritérii pro léčbu fingolimodem u nás byli do tohoto sledování obdobně zahrnuti pacienti, kde byl lék použit jako eskalační druhá volba v léčbě vysoce aktivní RS. Zařazeno bylo více než 3900 nemocných. Bylo dosaženo signifikantní redukce ročního počtu relapsů bez ohledu na typ léčiva předchozí léčby první volby. Taktéž bylo pozorováno alespoň šestiměsíční zlepšení skóre EDSS u 16 % pacientů, progrese EDSS u 10,3 % pacientů a stabilní hodnota skóre EDSS u 73,3 % pacientů v průběhu ročního sledování. Nežádoucí účinky vedly k ukončení léčby ve 3,8 % případů.
Dimethyl fumarát
Dimethyl fumarát (DMF) je nový perorální lék pro léčbu pacientů s RR RS. Lék je od března 2013 registrován v USA, dále postupně v Kanadě, Austrálii a od března 2014 také v Evropské unii. V době konání AAN bylo DMF léčeno celosvětově již více než 65 tisíc pacientů, bohužel čeští pacienti musí opět z důvodu extrémně dlouhých úhradových jednání čekat ještě minimálně do počátku roku 2015, než pro ně bude tento zajímavý lék také dostupný.DMF prokázal v klinických studiích efekt na redukci počtu relapsů, klinické invalidity i vývoje MR lézí ve srovnání s placebem, při dobrém bezpečnostním profilu. Z nežádoucích účinků je na prvním místě možná gastrointestinální intolerance po zahájení léčby (řada prací na AAN se zabývala právě tímto tématem s doporučením velmi pozvolné titrace při zahájení, která může toleranci na počátku léčby DMF zlepšit). Mezi časté nežádoucí reakce patří dále přetrvávající kožní zarudnutí, které nastupuje většinou za 2–3 hodiny po podání léku, a zdá se, že nebude zásadním důvodem pro ukončení léčby. U řady pacientů dochází také k lymfopenii, jež většinou není klinicky významná a také nevede k ukončení terapie.
Kolektiv autorů ze čtyř pracovišť v USA vedený Dr. Vissia Viglietta prezentoval bezpečnostní data šestiměsíční otevřené studie s přidaným DMF k léčbě zavedenými chorobu modifikujícími léčivy (disease modifying drugs, DMD) interferonem beta a glatiramer acetátem. Všichni pacienti, kterým byl k léčbě přidán DMF, prodělali alespoň jednu ataku v průběhu předcházejících dvanácti měsíců léčby DMD nebo měli průkaznou alespoň jednu novou aktivní lézi na MR v posledních šesti týdnech. Dle dat získaných u více než 100 pacientů nebyl prokázán vyšší výskyt nežádoucích účinků ve srovnání se známými nežádoucími účinky léčby jednotlivými přípravky v monoterapii. Uvedená data ukazují na možnost bezpečného převedení pacientů v případě aktivity RS z terapie DMD na podávání DMF bez nutnosti přerušení léčby.
Laquinimod
Prof. Giancarlo Comi z italského Milána prezentoval data z integrované analýzy dvou velkých klinických studií fáze III u pacientů s RR RS. V původní analýze prokázal laquinimod pozitivní účinek na vývoj tříměsíční potvrzené progrese současně s pozitivním ovlivněním vývoje mozkové atrofie vůči placebu. V této post hoc analýze byl zkoumán vývoj invalidity potvrzené nejen po třech měsících, ale i po šesti, devíti a dvanácti měsících. Také při zařazení těchto silnějších parametrů potvrzené progrese prokázal laquinimod pozitivní efekt. Při následující subanalýze, kde byli zvlášt zkoumáni pacienti s relapsy oproti pacientům s progresí bez relapsů, u kterých lze předpokládat vyšší stupeň neurodegenerace, prokázal laquinimod rovněž pozitivní efekt i u skupiny pacientů s progresí bez relapsu. Na základě těchto výsledků se zdá, že lék má potenciál pro ovlivnění primárně progresivní a sekundárně progresivní RS, a doufejme, že se v blízké době dočkáme klinické studie pro tyto pacienty. Bohužel zatím lék nemá registraci ani v USA, ani v EU.
Monoklonální protilátky
Natalizumab
Natalizumab představuje dosud nejúčinnější lék v terapii nemocných s RR RS. Na AAN byla prezentována řada prací dokládajících tuto mimořádnou efektivitu jak z hlediska klinického, tak z pohledu MR. Problémem u tohoto léčiva nicméně zůstává riziko rozvoje závažné oportunní infekce – progresivní multifokální leukoencefalopatie (PML), které je s podáváním natalizumabu spojeno. U pacientů s vysokým rizikem PML (léčba natalizumabem delší než 24 měsíců, vysoký titr protilátek proti JC viru v séru, předchozí léčba imunosupresivy) je indikována změna léčby, kdy v této situaci je v současnosti nejčastěji užívaným přípravkem fingolimod. Bohužel je tato změna léčby často provázena rizikem obnovení aktivity RS. Dr. Adil Maarouf a kol. vyhodnotili celkem 16 pacientů léčených nejprve alespoň šest měsíců natalizumabem a poté alespoň šest měsíců fingolimodem. Z uvedené skupiny došlo u devíti pacientů k reaktivaci RS po změně léčby. V pěti případech se jednalo o rebound onemocnění během prvních tří měsíců od změny léčby. Jediným prediktorem reaktivace RS po změně léčby z natalizumabu na fingolimod byla aktivita na MR před zahájením léčby natalizumabem.
Vzhledem k výjimečné účinnosti natalizumabu u pacientů s RS je samozřejmé, že se výzkumníci na celém světě snaží najít validní biomarkery, které by mohly pomoci ve vytipování pacientů s rizikem PML, a naopak umožnily pokračovat v léčbě pacientům, u kterých je riziko zcela minimální. Mezi tyto markery lze již dnes řadit stanovení titru protilátek proti JC viru v séru. Nově diskutovaným markerem je molekula CD62 na povrchu lymfocytů, patřící do rodiny selektinů. Kombinací obou markerů by mohlo být zřejmě dosaženo lepší predikce rizika PML, problémem však zatím bohužel zůstává otázka metodiky vyšetřování znaku CD62. Rovněž zřejmě nebude tento algoritmus stejně citlivý v predikci PML u pacientů, kteří byli předléčeni imunosupresivy.
Alemtuzumab
Alemtuzumab, monoklonální protilátka proti znaku CD52 na povrchu lymfocytů, prokázal vysokou účinnost v léčbě pacientů s RR RS ve dvou velkých klinických studiích fáze III – CARE-MS I (zařazeno 581 pacientů dosud zcela neléčených) a CARE-MS II (667 pacientů, u nichž selhala léčba léky první volby). V obou případech byl aktivním komparátorem interferon beta-1a podávaný 3krát týdně podkožně. Na základě výsledků byl lék na podzim 2013 registrován v Evropsk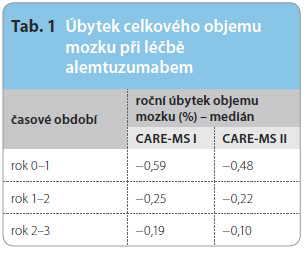 é unii. Dr. Arnold Douglas z Montrealu prezentoval tříletá MR data získaná z prodloužené fáze původní studie. Také ve třetím roce sledování byla většina pacientů bez známek nové MR aktivity, navíc měření vývoje mozkové atrofie prokázalo velmi zajímavé výsledky – úbytek objemu mozku se dostal do rozmezí úbytku, který vídáme u věkově odpovídající zdravé populace (tab. 1). Způsob podání alemtuzumabu je přitom pro pacienta velmi příjemný, jedná se o cyklus pěti infuzí během prvního roku a tří infuzí během druhého roku, dále „dle potřeby“, což je definováno novou klinickou aktivitou choroby (relaps) nebo MR aktivitou (alespoň dvě nové léze na MR). V prodloužené fázi původních studií dostalo ve třetím roce léčby třetí cyklus alemtuzumabu 18 % pacientů ze studie CARE-MS I a 20 % pacientů ze studie CARE-MS II.
é unii. Dr. Arnold Douglas z Montrealu prezentoval tříletá MR data získaná z prodloužené fáze původní studie. Také ve třetím roce sledování byla většina pacientů bez známek nové MR aktivity, navíc měření vývoje mozkové atrofie prokázalo velmi zajímavé výsledky – úbytek objemu mozku se dostal do rozmezí úbytku, který vídáme u věkově odpovídající zdravé populace (tab. 1). Způsob podání alemtuzumabu je přitom pro pacienta velmi příjemný, jedná se o cyklus pěti infuzí během prvního roku a tří infuzí během druhého roku, dále „dle potřeby“, což je definováno novou klinickou aktivitou choroby (relaps) nebo MR aktivitou (alespoň dvě nové léze na MR). V prodloužené fázi původních studií dostalo ve třetím roce léčby třetí cyklus alemtuzumabu 18 % pacientů ze studie CARE-MS I a 20 % pacientů ze studie CARE-MS II.
Důležitým momentem je bezpečnost léčby alemtuzumabem. I v extenzi obou studií se potvrdil výskyt autoimunitních komplikací asi u jedné třetiny pacientů. Jde zejména o poruchy štítné žlázy, podstatně méně často pak o případy autoimunitně podmíněné trombocytopenie a nefropatie. Důležité je, že ve většině případů jsou tyto nežádoucí účinky dobře řešitelné, pokud jsou diagnostikovány včas. Podmínkou léčby alemtuzumabem tedy nepochybně bude dlouhodobý (trvající nejméně 4 roky po podání 2. pulzu) a pečlivý monitoring, který bude vyžadovat velmi dobrou spolupráci pacienta.
Ofatumumab
Dr. Amit Bar-Or a kol. prezentovali pilotní data dvojitě slepé, placebem kontrolované studie MIRROR se subkutánním lékem ofatumumabem. Nadějná účinnost monoklonálních protilátek proti znaku CD20 exprimovanému B-lymfocyty u RS je již známa (u intravenózně podávaného ocrelizumabu aktuálně probíhá s českou účastí klinická studie fáze III, u intravenózně podávaného ofatumumabu se v letech 2008–2011 uskutečnila s českou účastí klinická studie fáze II). Také plně humánní subkutánně podávaná protilátka ofatumumab bude podle předložených výsledků i nadále zkoušena v rozsáhlejších studiích u pacientů s RS, protože ve výsledcích hodnocení fáze II byl lék v průběhu 12týdenního sledování spojen s redukcí počtu nových aktivních lézí na MR o 90 % a více ve srovnání s placebem (v dávkách 30 mg každých 12 týdnů, 60 mg každých 12 týdnů nebo 60 mg každý měsíc subkutánně). Nebyly pozorovány žádné případy oportunních infekcí včetně PML.
Novinky v etiopatogenezi RS
Pozornost byla věnována možnosti integrace již známých genetických a environmentálních faktorů s cílem vytvořit komplexní skóre, které by bylo schopno predikovat vznik nemoci a její tíži. Mezi genetickými faktory je to zejména lokus HLA-DRB*1501. Faktory prostředí představuje kouření, hladina protilátek proti viru Epsteina-Barrové v séru, sérová koncentrace vitaminu D, případně expozice slunečnímu záření. Diskutována byla také otázka vlivu obezity a hormonální antikoncepce. V případě většiny těchto faktorů jde o ovlivnitelné riziko ve vztahu k RS. Typickým příkladem je kouření; čím dál více se ukazuje, že negativní vliv kouření je z velké části způsoben vlivem na bronchiální slizniční imunitu, kde dochází k působení toxinů z cigaret na lymfocyty, které odtud mohou migrovat přímo do centrálního nervového systému.
Obdobně je tomu v případě střevní sliznice, která představuje jeden z nejvýznamnějších imunitních orgánů v těle. Problematice střevního mikrobiomu se dlouhodobě věnuje tým Prof. Howarda Weinera z Bostonu. Na AAN prezentovali autoři z tohoto vědeckého pracoviště výsledky práce, ve které bylo zkoumáno, zde existuje odlišnost ve složení střevního mikrobiálního ekosystému u pacientů s RS ve srovnání s tzv. zdravými kontrolami. U obou skupin byly vyšetřeny vzorky stolice a periferní krve a byly hledány korelace a odlišnosti mezi jednotlivými parametry. Zajímavým zjištěním byl významně vyšší výskyt Methanobrevibacter smithii v mikrobiomu pacientů s RS. Tato bakterie přitom může aktivovat imunoproliferativní a humorální odpověd imunitního systému. Práce pokračuje na větším vzorku pacientů s cílem zkoumat, zda a jak tyto bakterie ovlivňují imunitní buňky.
Význam vitaminu D podpořil Dr. Alberto Ascherio s kolektivem autorů v práci, která prezentovala data o možném prediktivním významu stanovení koncentrace vitaminu D v séru u pacientů s klinicky izolovaným syndromem (clinically isolated syndrome, CIS), kteří byli léčeni interferonem beta-1b v původní studii BENEFIT. Hladina 25-hydroxyvitaminu D byla u 216 pacientů s CIS hodnocena při vstupu do studie, dále po 6, 12 a 24 měsících sledování a korelována s klinickým a MR vývojem aktivity onemocnění v následujících pěti letech. Bylo prokázáno, že vstupní nízká koncentrace vitaminu D (≤ 50 nmol/l) úzce korelovala s pravděpodobností konverze CIS do klinicky definitivní RS stejně jako s počtem nových lézí na MR. Čím později byla léčba interferonem beta u pacientů s CIS zahájena, tím silnější byly uvedené korelace mezi nízkou vstupní hladinou vitaminu D a aktivitou onemocnění. Ze studie vyplynula vhodnost dalšího výzkumu vlivu nedostatku vitaminu D na prognózu RS a přídavné substituční léčby vitaminem D k již zavedené terapii léky modifikujícími průběh RS.
Práce byla podpořena programem PRVOUK-P26/LF1/4 a grantem NT13237-4/2012.