Antifosfolipidový syndrom – klinické projevy, diagnostika a možnosti léčby, komplikace
Souhrn:
Antifosfolipidový syndrom patří k nejdůležitějším trombofilním stavům. Je definován současnou přítomností klinické manifestace, to znamená trombózou nebo reprodukční ztrátou, a detekcí antifosfolipidových protilátek. Nicméně může být provázen i širokým spektrem tzv. „ne‑kritéria“ manifestací, jako jsou choroba srdečních chlopní, nefropatie, trombocytopenie, postižení kůže a jiné. Diagnóza antifosfolipidového syndromu je potvrzena naplněním diagnostických kritérií, která jsou definována mezinárodním konsensem. Hlavním léčebným přístupem je antitrombotická terapie. Imunomodulační léky však mohou hrát důležitou roli v některých situacích, zejména u „ne‑kritéria“ projevů. Nejzávažnější komplikací tohoto syndromu je jeho katastrofická manifestace. Tento život ohrožující stav je třeba mít na mysli u pacientů s antifosfolipidovými protilátkami, neboť vyžaduje časnou diagnostiku a intenzivní léčbu.
Key words: antiphospholipid syndrome – clinical manifestation – diagnosis – treatment – complications.
Summary:
Antiphospholipid syndrome represents one of the most important thrombophilic conditions. It is defined by simultaneous presence of clinical manifestation, i.e. thrombosis or reproductive failure, and detection of antiphospholipid antibodies. Nevertheless, it can be also accompanied by wide spectrum of so called non-criteria manifestations like heart valve disease, nephropathy, thrombocytopenia, skin involvement, and others. Diagnosis of the antiphospholipid syndrome is confirmed by fulfilling of diagnostic criteria, which are defined by international consensus. The main therapeutic approach is the antithrombotic treatment. However, immunomodulatory drugs can play an important role in some situations, especially in non-criteria manifestations. The most serious complication of this syndrome is catastrophic antiphospholipid syndrome. This life treating condition should be kept in mind in patients with antiphospholipid antibodies, as it needs early diagnosis and intensive treatment.
Úvod
Antifosfolipidové protilátky (antiphospholipid antibodies, APA) jsou velmi heterogenní skupinou autoprotilátek s různým významem pro své nositele. Některé z nich jsou prokazovány u zcela zdravých jedinců a spekuluje se o jejich významu v přirozených pochodech, jako je např. odstraňování reziduí apoptotického procesu. Část z těchto autoprotilátek se však nepochybně podílí na klinických projevech, které jsou asociovány s antifosfolipidovým syndromem (antiphospholipid syndrome, APS), a je zmiňována tzv. teorie druhého úderu. Velmi recentně byla definována kritéria patogenity APA, k nimž patří výskyt u většiny nemocných s APS, indukce humorální či buněčné odpov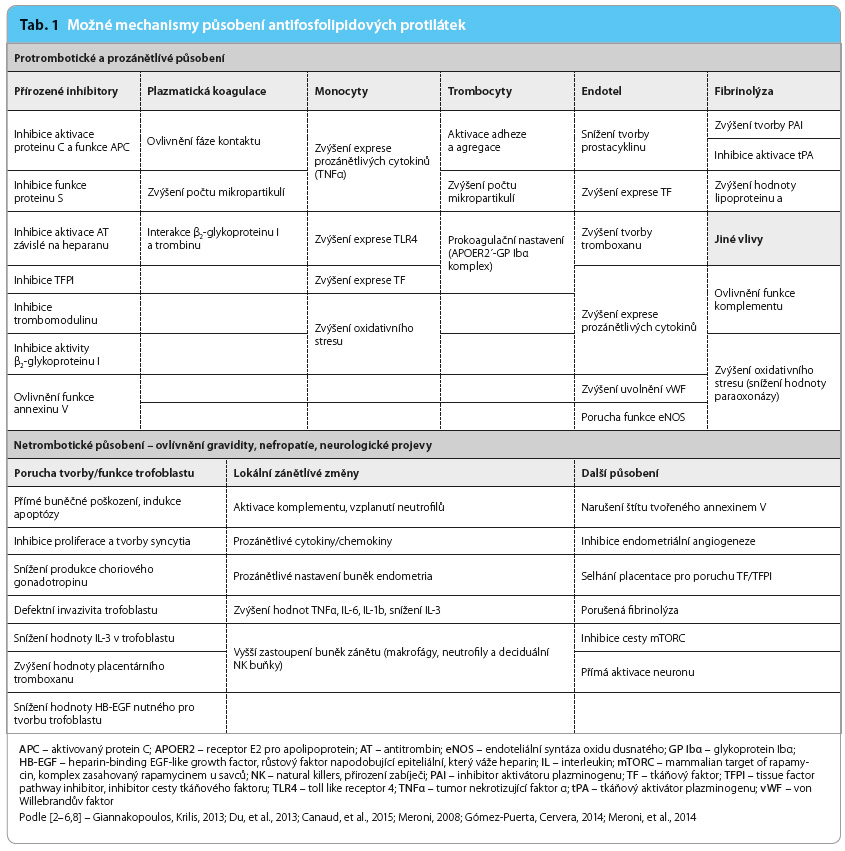 ědi kompatibilní s klinickou manifestací APS, indukce klinických projevů in vitro či na animálních modelech a v neposlední řadě i asociace s klinickou manifestací u pacientů s APS [1]. Nejvíce akceptovatelným patofyziologickým mechanismem působení APA je indukce protrombotického či prozánětlivého stavu [2–6]. Toto se může odehrávat na více úrovních procesu krevního srážení, resp. zánětu. Nicméně relativně nedávná publikace upozornila na to, že se při některých klinických projevech může uplatnit i vznik vaskulopatie spojené s intimální hyperplazií, jež je indukována cestou signálních drah AKT/mTORC (proteinkináza B/savčí komplex zasahovaný rapamycinem), což je následováno proliferací okolních hladkých svalových buněk cév [4]. Další účinky směřující k přímému ovlivnění funkčního stavu buněk či tkání se uplatňují v případě působení APA zejména u těhotenských komplikací APS nebo jsou předpokládány u některých neurologických projevů, kdy se počítá s přímým ovlivněním neuronu. Přehled možných vlivů APA je uvedenv tab. 1 [2–6].
ědi kompatibilní s klinickou manifestací APS, indukce klinických projevů in vitro či na animálních modelech a v neposlední řadě i asociace s klinickou manifestací u pacientů s APS [1]. Nejvíce akceptovatelným patofyziologickým mechanismem působení APA je indukce protrombotického či prozánětlivého stavu [2–6]. Toto se může odehrávat na více úrovních procesu krevního srážení, resp. zánětu. Nicméně relativně nedávná publikace upozornila na to, že se při některých klinických projevech může uplatnit i vznik vaskulopatie spojené s intimální hyperplazií, jež je indukována cestou signálních drah AKT/mTORC (proteinkináza B/savčí komplex zasahovaný rapamycinem), což je následováno proliferací okolních hladkých svalových buněk cév [4]. Další účinky směřující k přímému ovlivnění funkčního stavu buněk či tkání se uplatňují v případě působení APA zejména u těhotenských komplikací APS nebo jsou předpokládány u některých neurologických projevů, kdy se počítá s přímým ovlivněním neuronu. Přehled možných vlivů APA je uvedenv tab. 1 [2–6].
Klinické projevy
Antifosfolipidový syndrom je klinicko laboratorní jednotka, která je provázena klinickými projevy ve formě trombózy či přesně definované reprodukční ztráty (viz dále) a přítomností APA. Proto je zřejmé, že trombóza bude nejčastější klinickou manifestací. Dle studie Euro Phospholipid project, do níž bylo zařazeno 1 000 pacientů s APS [7], představovala nejčastější klinickou manifestaci hluboká žilní trombóza, která postihuje dle této kohorty 38,9 % nemocných; v literatuře je udávána tato klinická manifestace u 29–55 % nositelů APA, plicní embolie je u nemocných s APS diagnostikována ve 14,1 %. Ve skupině nemocných s prodělanou hlubokou žilní trombózou je četnost diagnózy APS odhadována na 9,5 %. Nicméně u nemocných s APS může trombóza vzniknout v kterékoliv části cévního řečiště a nejsou vzácností trombózy v abnormálních lokalizacích. Postižení tepenného řečiště se nejčastěji manifestuje ve formě cévní mozkové příhody (19,8 %) či transitorní ischemické ataky (11,1 %), zatímco v populaci nemocných s ischemií v oblasti centrální nervové soustavy se APA prokazují ve 13 % případů. Méně časté je dle Euro Phospholipid project postižení koronárního řečiště (5,5 %), nicméně pozitivita APA u pacientů s infarktem myokardu je odhadována na 11 %. Mikrotrombotizace v nejrůznějších tkáních či orgánech může být příčinou komplikací kardiálních (syndrom X), kožních (pseudovaskulitické léze, kožní či digitální gangréna), endokrinních (nejčastěji selhání nadledvinek), ale mohou se uplatnit i postižení reprodukčních či jiných endokrinních orgánů, resp. postižení v gastrointestinální oblasti (jícnová či mezenteriální ischemie, splenický infarkt). Postižení oka může být příčinou amaurosis fugax či se projeví jako trombóza retinální tepny.
Kromě klinických projevů, které mohou být jasně vysvětleny vznikem trombózy či mikrotrombózy v určité části cirkulace, se u nemocných s APS setkáváme s poměrně širokým spektrem manifestací, které nelze trombózou, a dokonce ani mikrotrombotizací tkáně uspokojivě vysvětlit, resp. u nichž je toto vysvětlení nejisté. K těmto patří projevy neurologické – migréna je jako jedna z nejčastějších „ne kritéria“ manifestací (popisována u 19,8 % nemocných s APA), dále epilepsie (7 %) a chorea (1,3 %). K závažným neurologickým projevům patří porucha kognitivních funkcí, která u nemocných bez systémového lupus erythematodes (SLE) může postihovat až 38 % nositelů APA. K dalším těmto manifestacím patří postižení kardiovaskulární – zde je nutno zmínit především postižení chlopní (11,6 %) či kardiomyopatii (2,9 %). Kůže je taktéž relativně častým místem klinických projevů netrombotických příčin (livedo reticularis či livedo racemosa – 24,1 %, může se vyskytnout i atrophia blanche). Nálezy hematologické jsou diagnostikovány ve formě autoimunitní hemolytické anémie (9,7 %) či trombocytopenie (29,6 %). K zajímavým a nepříliš vzácným postižením patří avaskulární kostní nekróza (2,4 %), která postihuje nejčastěji metatarsy. Patofyziologicky heterogenní je nejspíše nefropatie asociovaná s APA, která postihuje více než třetinu nemocných [8]. Část nálezů je zapříčiněna stenózou renální tepny, nicméně může se objevit i netrombogenní vaskulopatie postihující malé arterie, arterioly či glomerulární kapiláry, kdy se více uplatňuje hypertrofie intimy cév [4]. Postižení ledvin je u APS provázeno klinickou manifestací ve formě hypertenze a/nebo proteinurie, nicméně přesvědčivá diagnostika nefropatie asociované s APA je pouze histologická, kdy je nutno naplnit dříve definovaný morfologicko patologický obraz a vyloučit zánětlivé postižení cévy.
Další významnou klinickou manifestací, která je jasně asociovaná s přítomností APA, jsou poruchy reprodukce. Prevalence APA je v případě samovolných potratů udávána v širokém rozmezí od 5 % do 50 % (průměrně 15,5 %) [9], u žen se ztrátou plodu po 20. týdnu gravidity dosahuje prevalence až 30 %. Příčina heterogenity těchto nálezů tkví ve špatné standardizaci laboratorních diagnostických metod a v nejednotném nastavení limitních hodnot. V již zmiňovaném Euro Phospholipid project bylo analyzováno 1 580 těhotenství u 590 žen, kdy časné ztráty byly zjištěny u 35,4 % gravidit, pozdní u 16,9 % a jen 47,7 % těhotenství bylo ukončeno narozením životaschopného plodu. K dalším komplikacím gravidity, které jsou asociovány s nálezem APA, patří preeklampsie (9,5 %), eklampsie (4,4 %) a abrupce placenty (2,0 %) a předčasný porod (10,6 %) [6]. Poněkud více kontroverzní je souvislost mezi primární infertilitou či selháním technik asistované reprodukce a výskytem APA. Ačkoliv některé nálezy z animálních modelů připouštějí teoretickou asociaci, která, zdá se, vyplývá i z běžné klinické praxe, metaanalýza tyto vztahy nepotvrdila; v případě selhání fertilizace in vitro preimplantační diagnostika zjistila u 60 % embryí chromosomální aberace [10].
Podle klinické manifestace jsou rozlišovány některé další podjednotky, jako je například rychle probíhající APS či APS spojený s trombotickou mikroangiopatií, případně i tzv. séronegativní APS, kdy jsou klinické projevy prakticky totožné, avšak přítomnost diagnostických APA byla opakovaně vyloučena, resp. byla vyloučena příčina jejich dočasné negativity [11]. Přehled klinických projevů přináší tab. 2.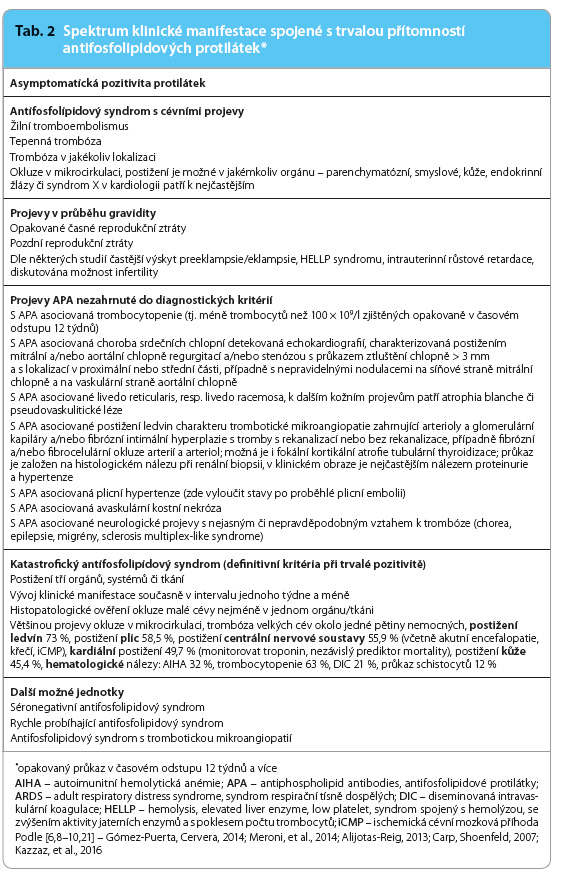
Diagnostika
Diagnóza APS je stanovena naplněním diagnostických kritérií. Tato diagnostická kritéria byla navržena počátkem devadesátých let, opakovaně byla revidována a jejich poslední a doposud platná verze vznikla na jedenáctém Mezinárodním kongresu o antifosfolipidových protilátkách v Sydney v roce 2004 [12]. Přehled diagnostických kritérií podává tab. 3. K naplnění diagnózy APS je nezbytný průkaz jednoho klinického a jednoho laboratorního kritéria, přičemž časový odstup mezi klinickou manifestací a průkazem APA je možný v nejdelším intervalu pěti let. Zatímco stanovení 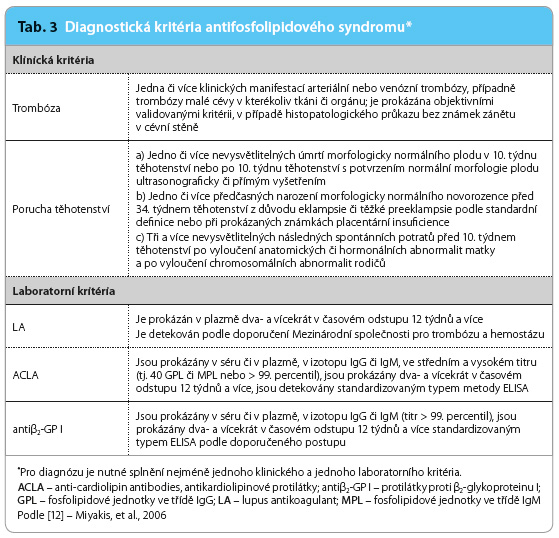 přítomnosti či nepřítomnosti klinických kritérií je v naprosté většině případů relativně jednoduché a využívá nejčastěji jen standardního vyšetření pacienta, je průkaz APA relativně nesnadný, a to přes veškerý pokrok laboratorní diagnostiky. Antifosfolipidové protilátky stanovuje řada laboratoří, nicméně v běžné praxi se relativně často setkáváme se situací, kdy pozitivní nález zjištěný v jedné laboratoři se v jiné laboratoři nepotvrdí. Celou problematiku laboratorní diagnostiky je nutno rozdělit na dva odlišné typy vyšetření, a to na průkaz lupus antikoagulant, tedy vyšetření na bázi testů krevního srážení, a na diagnostiku sérologickou – průkaz protilátek proti kardiolipinu a proti β2 glykoproteinu I pomocí testu ELISA (Enzyme Linked ImmunoSorbent Assay) nebo chemoluminiscencí.
přítomnosti či nepřítomnosti klinických kritérií je v naprosté většině případů relativně jednoduché a využívá nejčastěji jen standardního vyšetření pacienta, je průkaz APA relativně nesnadný, a to přes veškerý pokrok laboratorní diagnostiky. Antifosfolipidové protilátky stanovuje řada laboratoří, nicméně v běžné praxi se relativně často setkáváme se situací, kdy pozitivní nález zjištěný v jedné laboratoři se v jiné laboratoři nepotvrdí. Celou problematiku laboratorní diagnostiky je nutno rozdělit na dva odlišné typy vyšetření, a to na průkaz lupus antikoagulant, tedy vyšetření na bázi testů krevního srážení, a na diagnostiku sérologickou – průkaz protilátek proti kardiolipinu a proti β2 glykoproteinu I pomocí testu ELISA (Enzyme Linked ImmunoSorbent Assay) nebo chemoluminiscencí.
Průkaz lupus antikoagulant
Lupus antikoagulant představuje časově nezávislý inhibitor krevního srážení, který prodlužuje hemokoagulační testy závislé na fosfolipidech, nejčastěji aktivovaný parciální tromboplastinový test (aPTT). Jeho průkaz je relativně náročný a závislý i na preanalytických podmínkách, jako je např. způsob odběru a časné zpracování bezdestičkové plazmy speciální centrifugací. Z tohoto důvodu může být interpretace testů prováděných ve vzdálené laboratoři velmi komplikovaná, včetně relativně vysoké možnosti jak falešně pozitivních, tak i falešně negativních nálezů. I vlastní analytický proces je dosti komplikovaný, využívá více testů a více kroků potvrzení přítomnosti inhibitoru – testy screeningové a směsné, resp. testy průkazu jeho fosfolipidové závislosti – tedy testy konfirmační. Aby se pokryla možnost zásahu lupus antikoagulant na různých úrovních krevního srážení, měly by být k diagnostice použity nejméně dva typy testů, jednak modifikovaný aPTT se sníženým obsahem fosfolipidů, jednak test s jedem Russelovy zmije (dilute Russell‘s viper venom time, dRVVT). Typ průkazu – jen na bázi aPTT, jen na bázi dRVVT či oba pozitivní – pak má diagnostický význam, neboť v tomto pořadí pozitivity narůstá i míra rizika trombózy. Celý diagnostický proces se řídí mezinárodními doporučeními, včetně nutnosti nastavení rozhraní pozitivního a negativního nálezu za standardních podmínek v každé laboratoři [13–16].
Odběr krve na průkaz lupus antikoagulant by neměl být prováděn v průběhu akutního onemocnění pro vysoké riziko falešně pozitivních nálezů. Jeho stanovení je významně ovlivněno podávanou léčbou, zejména nefrakcionovaným heparinem, ale i terapeutickými dávkami nízkomolekulárních heparinů, a vůbec není doporučováno při léčbě přímými perorálními antitrombotiky typu dabigatranu, apixabanu či rivaroxabanu pro vysoké riziko falešně pozitivních nálezů [16]. Při izolovaném průkazu lupus antikoagulant bez sérologického potvrzení jiných APA by se mělo vždy přihlédnout ke komplexnímu klinicko laboratornímu posouzení.
Sérologická diagnostika protilátek
Sérologický průkaz protilátek podmiňujících APS je zdánlivě jednoduchý (ELISA nebo chemoluminiscence), nicméně i zde svou roli hraje neoptimální standardizace a nutnost nastavení vlastních „cut off“ kritérií v každé laboratoři [15]. V běžné praxi se nezřídka setkáváme se situací, že to, co je v jedné laboratoři pozitivní, je v jiné negativní. Každý lékař, který využívá laboratorní diagnostiku APA, by měl mít náležitou informaci o tom, jaké nastavení pozitivních a negativních nálezů používá laboratoř, která s ním spolupracuje, včetně zapojení do systému externí kontroly kvality [13–16].
Dalším diagnostickým problémem je skutečnost, že byly prokázány významné klinicko patofyziologické souvislosti s řadou autoprotilátek, které sice nejsou akceptovány jako diagnostické pro stanovení APS, nicméně nelze vyloučit jejich důležitost pro manifestaci klinických projevů spojených s APS. Zde se v prvé řadě nabízí třída imunoglobulinů A (IgA) – „klasických“ APA, tedy protilátek proti kardiolipinu, resp. proti β2 glykoproteinu I. Tyto jsou zmiňovány zejména v souvislosti s poruchami koncepce či s neurologickými projevy navozenými ischemií. Svou roli mohou hrát i protilátky detekované jako protilátky proti annexinu V, proti vimentinu, proti fosfatidylethanolaminu či proti komplexu fosfatidyl serin protrombin [8]. V současné době je diskutována taktéž problematika průkazu protilátek proti doméně I β2 glykoproteinu I. Tyto protilátky se zdají být zřetelně více asociovány s klinickými projevy APS než protilátky proti jiným doménám této molekuly (zejména proti doméně IV či V), které se jeví jako netrombogenní [8].
Při diagnostickém procesu musí být věnována pozornost i skutečnosti, zda je prokázán jen jeden typ APA, tedy tzv. typ II laboratorních diagnostických kritérií, nebo je pozitivních autoprotilátek více, tj. pozitivita typu I [12]. Z klinického pohledu nejvýznamnější se zdá být tzv. trojnásobná pozitivita, tedy přítomnost jak lupus antikoagulant, tak vysokých titrů protilátek proti kardiolipinu i proti β2 glykoproteinu I ve třídě imunoglobulinů G (IgG). Nositelé této kombinace autoprotilátek jsou v dlouhodobém sledování ohroženi nejvyšším rizikem vzniku trombózy [17].
Profylaxe a léčba nemocných s antifosfolipidovými protilátkami
Doporučení, jak zabránit vzniku klinických projevů APS u nositelů APA, resp. jak je léčit v případě již zřejmé klinické manifestace, byla publikována před pěti lety [18], nicméně v některých situacích nejsou jednoznačná. 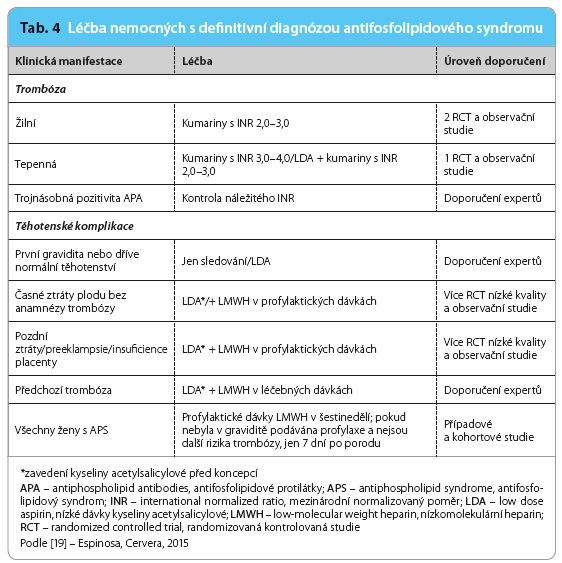 Přehled současných názorů předkládá tab. 4 a 5 [19]. Všem pacientům, u nichž byla prokázána trvalá pozitivita APA, je doporučena přísná kontrola dalších kardiovaskulárních rizik a zabezpečení antitrombotické profylaxe v situacích, které riziko vzniku trombózy dále zvyšují; stejný postup se doporučuje i u jiných trombofilních stavů. K těmto situacím je nutno počítat operační výkony, déletrvající imobilizaci, ovariální stimulaci, těhotenství, porod a šestinedělí. Lékem volby je v těchto případech dočasně podávaný nízkomolekulární heparin v profylaktických dávkách. Dále je nutno rozlišovat, zda APA byly prokázány primárně, tedy bez vyvolávajícího onemocnění, zejména SLE, nebo zda naopak jde o nemocného, u nějž byl SLE již dříve diagnostikován. V prvním případě je primární profylaxe zvažována, jde li o profil APA s vysokým rizikem trombózy (tj. lupus antikoagulant, vysoké titry sérologicky detekovaných APA ve třídě IgG, resp. kombinace obou, zejména trojnásobná pozitivita), a zvláště v případech, kdy jsou přítomny i další rizikové faktory trombózy a/nebo je jasně pozitivní anamnéza těhotenských komplikací [20]. Lékem volby je kyselina acetylsalicylová v dávkách 75–100 mg denně (low dose aspirin, LDA) [17]. U nemocných se SLE je doporučováno pravidelně pátrat po přítomnosti APA, těm z pacientů, u nichž je opakovaně prokázán lupus antikoagulant a/nebo střední až vysoké titry protilátek proti kardiolipinu či proti β2 glykoproteinu I, je doporučeno v primární profylaxi podávat hydroxychlorochin a LDA [18]. I zde je nutno zvažovat posílení antitrombotické profylaxe nízkomolek
Přehled současných názorů předkládá tab. 4 a 5 [19]. Všem pacientům, u nichž byla prokázána trvalá pozitivita APA, je doporučena přísná kontrola dalších kardiovaskulárních rizik a zabezpečení antitrombotické profylaxe v situacích, které riziko vzniku trombózy dále zvyšují; stejný postup se doporučuje i u jiných trombofilních stavů. K těmto situacím je nutno počítat operační výkony, déletrvající imobilizaci, ovariální stimulaci, těhotenství, porod a šestinedělí. Lékem volby je v těchto případech dočasně podávaný nízkomolekulární heparin v profylaktických dávkách. Dále je nutno rozlišovat, zda APA byly prokázány primárně, tedy bez vyvolávajícího onemocnění, zejména SLE, nebo zda naopak jde o nemocného, u nějž byl SLE již dříve diagnostikován. V prvním případě je primární profylaxe zvažována, jde li o profil APA s vysokým rizikem trombózy (tj. lupus antikoagulant, vysoké titry sérologicky detekovaných APA ve třídě IgG, resp. kombinace obou, zejména trojnásobná pozitivita), a zvláště v případech, kdy jsou přítomny i další rizikové faktory trombózy a/nebo je jasně pozitivní anamnéza těhotenských komplikací [20]. Lékem volby je kyselina acetylsalicylová v dávkách 75–100 mg denně (low dose aspirin, LDA) [17]. U nemocných se SLE je doporučováno pravidelně pátrat po přítomnosti APA, těm z pacientů, u nichž je opakovaně prokázán lupus antikoagulant a/nebo střední až vysoké titry protilátek proti kardiolipinu či proti β2 glykoproteinu I, je doporučeno v primární profylaxi podávat hydroxychlorochin a LDA [18]. I zde je nutno zvažovat posílení antitrombotické profylaxe nízkomolek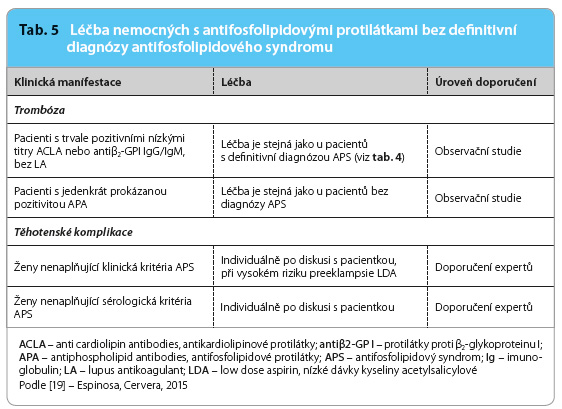 ulárními hepariny v rizikových situacích, které v porovnání s primárním výskytem APA zahrnují navíc akutní vzplanutí choroby, léčbu kožních lézí thalidomidem, nefrotický syndrom a/nebo infekce [20].
ulárními hepariny v rizikových situacích, které v porovnání s primárním výskytem APA zahrnují navíc akutní vzplanutí choroby, léčbu kožních lézí thalidomidem, nefrotický syndrom a/nebo infekce [20].
Akutní léčba trombotické manifestace APS ať již v žilním či v tepenném řečišti se nebude v ničem odlišovat od léčby ve všeobecné populaci, neboť často není k dispozici informace o perzistentní (tj. více než 12 týdnů trvající) pozitivitě APA. Nicméně došlo li ke klinické manifestaci u nemocných, u nichž je přítomnost APA dlouhodobě zjištěna, měli bychom v případě hluboké žilní trombózy či plicní embolie volit použití přímých perorálních antitrombotik cílených proti aktivovanému faktoru Xa (xabany), resp. proti aktivovanému faktoru II, velmi obezřetně. Tyto léky se ukázaly být nejméně stejně účinné a nejméně stejně bezpečné v uvedených indikacích ve všeobecné populaci, nicméně data o jejich účinnosti u APS doposud chybějí; je proto nutno vyčkat výsledků probíhajících studií, které budou ukončeny nejdříve v roce 2017 [19]. Tyto přípravky by tudíž měly být vyhrazeny pro nemocné s definitivně stanovenou diagnózou APS, kteří mají přecitlivělost na kumariny nebo je netolerují, případně je u nich obtížně nastavitelná náležitá hodnota INR (international normalized ratio, mezinárodní normalizovaný poměr). Tudíž je u definitivní diagnózy APS nutno počítat spíše s léčbou klasickou, tedy nízkomolekulární heparin následovaný kumariny u pacientů s venózní trombózou, kdy hodnota cílového INR je doporučena v klasickém rozmezí 2,0–3,0 [18,19]. V případě trombózy tepenné je situace mnohem kontroverznější, neboť stanovisko expertů pracovní skupiny, jak v těchto případech postupovat, se neshoduje. V sekundární profylaxi je doporučeno podávat jak léčbu kumariny s cílovým INR 3,0–4,0, tak i kombinaci klasicky dávkovaných kumarinů současně s LDA. U pacientů bez SLE, kteří mají APA s nízkým trombotickým profilem, by mohla být zvážena i klasická protidestičková léčba [18].
Každopádně je léčba trombózy u nemocných s APA zvažována jako doživotní, i když toto doporučení je založeno na nízké hladině kvality průkazu. U nemocných s jednorázovým průkazem APA, který nebyl ověřen opakovaným stanovením, je preferován běžný léčebný režim používaný u osob, u nichž pozitivita APA nikdy prokázána nebyla [19]. Navíc je nutno přihlédnout i k okolnostem, zda šlo o trombózu idiopatickou, či sekundární po působení dalšího vyvolávajícího a odstranitelného faktoru.
Antifosfolipidový syndrom a těhotenství
Léčba i profylaxe APS manifestujícího se v graviditě či „jen“ pozitivity APA je dominantně závislá na předchozí anamnéze. V případech jasného naplnění kritérií APS, ať již ve formě trombotických či těhotenských komplikací, je situace poměrně jasná a daná dříve publikovanými doporučeními [18,19], která řeší zhruba 70–80 % případů, viz. tab. 4 a 5. Kromě LDA, aplikovaného nejčastěji 3–4 týdny před koncepcí, může být použit nízkomolekulární heparin přidaný optimálně bezprostředně po zjištění gravidity, při užití technik asistované reprodukce a jasně naplněných kritérií APS bezprostředně po transferu.
Refrakterní případy
Léčba refrakterních případů je řešena individuálně. Recidiva trombózy není vzácná, v Euro Phospholipid project došlo během desetiletého sledování k této komplikaci u třetiny pacientů. Je doporučeno navýšení dávek kumarinu (INR > 3,0), pakliže k recidivě došlo při náležité úrovni antikoagulace, přidání LDA či hydroxychlorochinu ke stávající antikoagulační léčbě, zvážen může být i rituximab. K dalším lékům, jež mohou příznivě ovlivnit prozánětlivé a protrombotické nastavení endotelu navozené působením APA, patří statiny. Selhání léčby těhotenských komplikací vede nejčastěji k časnějšímu podání medikace (LDA více než 4 týdny před koncepcí), k navýšení dávek nízkomolekulárních heparinů, k přidání malých dávek kortikosteroidů nebo hydroxychlorochinu nejčastěji na období prvního trimestru. Zmiňováno je však i používání vysokodávkovaných intravenózních gamaglobulinů, případně i velkoobjemové plazmafarézy, resp. monoklonálních protilátek blokujících tumor nekrotizující faktor [9,19].
„Ne kritéria“ manifestace
Léčba směřující k ovlivnění imunitního systému s cílem zamezit tvorbě autoprotilátek je více využívána u „ne kritéria“ klinických manifestací APS. Hemolytická anémie či trombocytopenie asociovaná s APA jsou léčeny jako jiné imunitní cytopenie – kortikosteroidy, intravenózními imunoglobuliny, imunosupresivy, splenektomií či rituximabem, v případě nízkých počtů trombocytů bylo úspěšné i podávání agonistů trombopoetinového receptoru (eltrombopag) [19]. U nemocných s trombocytopenií je popisováno vyšší riziko vzniku trombózy, a proto těm pacientům, kteří při léčbě dosáhnou počtu destiček vyššího než 50 × 109/l a současně je u nich přítomen lupus antikoagulant jako nejvíce trombogenní APA nebo mají další vaskulární rizikové faktory, je přidávána léčba LDA nebo hydroxychlorochinem.
I u nemocných se závažným neurologickým postižením (transversální myelitida, choroba napodobující sclerosis multiplex) je k antikoagulační léčbě přidána terapie kortikosteroidy a imunosupresivy.
Pacienti s postižením chlopní jsou určeni k antikoagulační léčbě, jsou li jejich potíže provázeny klinickou manifestací, asymptomatickým nemocným je podávána LDA. Není u nich však všeobecně doporučována profylaxe infekční endokarditidy. Antitrombotická léčba však postižení chlopní obvykle nezmění a část pacientů musí podstoupit řešení operativní.
Nefropatie asociovaná s APA bude taktéž vyžadovat antitrombotickou medikaci, doporučena je jak protidestičková, tak i antikoagulační léčba. Všichni tito nemocní by měli mít náležitě kompenzovanou hypertenzi a proteinurii zavedením léčby inhibitory angiotensin konvertujícího enzymu a blokátory angiotensinových receptorů.
Komplikace
Nejzávažnější a nezřídka fatální komplikací APS je jeho „katastrofická manifestace“, která naštěstí postihuje necelé 1 % nemocných. Nicméně je provázena relativně vysokou úmrtností (48 %), která se však dle dostupných registrů snižuje úměrně tomu, jak se urychluje diagnostika a jak časně je zaváděna adekvátní intenzivní léčba – dnes umírá zhruba třetina nemocných, jedná li se o primární katastrofický antifosfolipidový syndrom (CAPS), tedy ne o APS provázející SLE, kde je prognóza nepříznivější (umírá 58 % nemocných). Proč tato komplikace vzniká, není z patofyziologického pohledu úplně jasné. Jistě se uplatňuje silný trombogenní potenciál navozený APA (viz tab. 1), resp. nerovnováha mezi generací trombinu a/nebo fibrinu a jeho lýzou, neboť část klinické manifestace je ovlivnitelná antitrombotickou terapií. Nicméně se předpokládá současné uplatnění i aktivace cytokinové kaskády a/nebo dochází k rozvoji syndromu systémové zánětlivé odpovědi (systemic inflammatory response syndrome, SIRS) [21]. Spekuluje se o úloze aktivovaného intracelulárního transkripčního faktoru (jaderný faktor kappa B a aktivace extracelulární komplementové kaskády), což jsou výhledově zajímavé cíle pro terapii. Mezi faktory vyvolávajícími CAPS jsou zmiňovány zejména infekce (E. coli, Klebsiella, Salmonella, Shigella, Staphylococcus, Streptococcus, herpesviry, HIV). Dále jsou to malignity, především hematologické, ale i karcinomy plic a tračníku, svou roli může hrát i těhotenství, vzplanutí SLE a nedostatečná antitrombotická terapie.
Katastrofický antifosfolipidový syndrom se manifestuje zejména mikrotrombotizací orgánů, což vede k jejich selhání, zatímco běžná hluboká žilní trombóza je spíše vzácná. Diagnostický algoritmus je založen na průkazu současného postižení tří orgánových systémů a tkání vznikajícího v časovém rozmezí jednoho týdne s histologickým průkazem postižení cévy trombózou nejméně v jednom z těchto případů (tab. 2), a to za současného průkazu nejméně jedné APA za standardních kritérií (tab. 3). Přestože průkaz lupus antikoagulant má s klinickou manifestací CAPS nejsilnější asociaci a je zjišťován u 82 % nemocných s touto diagnózou, je v akutním stavu pacienta ovlivněn skutečností, že může být prokázán v intenzivní péči bezmála u 50 % případů z jiných příčin (probíhající infekce, podávání katecholaminů, nádory), které jsou nezávislé na APS. Proto je v tomto případě vhodnější spoléhat se na sérologickou diagnostiku.
Nejzávažnějším momentem zjištění diagnózy CAPS je skutečnost, že až téměř polovina nemocných může mít CAPS jako první klinickou manifestaci APS, a proto je třeba mít tuto možnost na mysli při závažném klinickém stavu pacienta s multiorgánovým trombotickým postižením. V diferenciální diagnostice je pak zvažována zejména trombotická trombocytopenická purpura/hemolyticko uremický syndrom či jiné trombotické mikroangiopatie (zejména těžká preeklampsie‚ eklampsie či syndrom HELLP [hemolysis, elevated liver enzymes, and low platelets, hemolýza, elevace aktivity jaterních enzymů, snížený počet trombocytů] v graviditě či v časném puerperiu), diseminovaná intravaskulární koagulopatie, heparinem indukovaná trombocytopenie a renální krize u sklerodermie [21]. Pro další léčbu je nutno rozlišit sekundární CAPS u SLE.
Jak již bylo zmíněno, prognóza CAPS se odvíjí od časné diagnostiky a zahájení intenzivní léčby. Základem této je umístění pacienta na jednotce intenzivní péče, intenzivní léčba antitrombotická – nefrakcionovaný heparin nebo nízkomolekulární heparin, následně kumariny, případně fibrinolytika. Role protidestičkové léčby či přímých perorálních antitrombotik není v těchto případech vyjasněna. Další nepodkročitelnou léčbu představují kortikosteroidy. Ty jsou často používány intravenózně a relativně ve vysokém dávkování. Jde li o nemocné se SLE, pak je dalším terapeutickým přístupem cyklofosfamid; zde je však nutné mít jasnou diagnózu, neboť u pacientů s primárním CAPS není tato léčba přínosná. Přidání dalších terapeutických postupů, jako je výměnná plazmaferéza či vysokodávkované intravenózní imunoglobuliny, výrazně zlepšuje prognózu nemocných, zejména v případech, kdy je přítomna trombotická mikroangiopatie nebo kdy nedochází k jasné klinické odpovědi na standardní léčbu. V refrakterních případech či tam, kde se předpokládá významná aktivace komplementu, jsou ke zvážení ještě rituximab (monoklonální protilátka anti CD20) či eculizumab (humanizovaná monoklonální protilátka bránící aktivaci C5 složky komplementu).
Závěr
Péče o nemocné s APS je komplikovaná zejména v tom, že syndrom má velmi heterogenní klinickou manifestaci a počínaje lékařem intenzivní péče až po rutinního dermatologa se s takovýmto pacientem může setkat kterýkoliv lékař, hematology, gynekology, revmatology, kardiology, nefrology, pneumology či internisty nevyjímaje. Z tohoto pohledu je nezbytné, aby měl každý ošetřující lékař tuto diagnostickou jednotku na mysli, a to jak v rámci akutních, resp. i život ohrožujících situací, tak i chronických stabilizovaných stavů, které však vzbuzují podezření na APS. Diagnostika není jednoduchá, může být často „předimenzovaná“, což se může odrazit v neadekvátně intenzivní či zbytečně dlouhodobé terapii. Z tohoto pohledu je nezbytné dodržování stanovených diagnostických i léčebných standardů, případně soustředění specializované péče o zmíněné nemocné do center připravených na adekvátní diagnostiku a léčbu.
Seznam použité literatury
- [1] Lackner KJ, Müller‑Calleja N. Pathogenesis of the antiphospholipid syndrome revisited. Time to challenge the dogma. J Thromb Haemost 2016: 14: 1117–1120.
- [2] Giannakopoulos B, Krilis SA. The pathogenesis of the antiphospholipid syndrome. NEJM 2013; 368: 1033–1044.
- [3] Du VX, Kelchtermans H, de Groot PG, de Laat B. From antibody to clinical phenotype, the black box of the antiphospholipid syndrome: Pathogenic mechanisms of antiphospholipid syndrome. Thromb Research 2013; 132: 319–326.
- [4] Canaud G, Legendre C, Terzi F. AKT/mTORC pathway in antiphospholipid related vasculopathy: a new player in the game. Lupus 2015; 24: 227–230.
- [5] Meroni PL. Pathogenesis of the antiphospholipid syndrome: Additional example of the mosaic of automimmunity J Autoimmun 2008; 30: 99–103.
- [6] Gómez‑Puerta JA, Cervera R. Diagnosis and classification of the antiphospholipid syndrome. J Autoimmun 2014; 48–49: 20–25.
- [7] Cervera R, Boffa MC, Khamashta MA, Hughes GR. The Euro‑Phospholipid project: epidemiology of the antiphospholipid syndrome in Europe. Lupus 2009; 18: 889–893.
- [8] Meroni PJ, Chighizola CB, Rovelli F, et al. Antiphospholipid syndrome in 2014: more clinical manifestations, novel pathogenic players and emerging biomarkers. Arthr Research Therap 2014; 16: 209.
- [9] Alijotas‑Reig J. Treatment of refractory obstetric antiphospholipid syndrome: the state of the art and new trends in the therapeutic management. Lupus 2013; 22: 6–17.
- [10] Carp HJA, Shoenfeld Y. Anti‑phospholipid antibodies and infertility. Clin Rev Aller Immunol 2007; 32: 159–161.
- [11] Cervera R, Conti F, Doria A, et al. Does seronegative antiphospholipid syndrome really exist? Autoim Rev 2012; 11: 581–584.
- [12] Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006; 4: 295–306.
- [13] Pengo V, Tripodi A, Reber G, et al. Update of the guidelines for lupus anticoagulant detection. J Thromb Hemost 2009; 7: 1737–1740.