Defekt v SHOX genu – příčina familiárního malého vzrůstu
Insuficience SHOX genu je jednou z poměrně častých příčin geneticky podmíněné růstové retardace. SHOX gen je lokalizován v pseudoautozomální oblasti 1 (PAR1) obou pohlavních chromozomů (Xp22.3, Yp11.3). Jedná se o regulační gen kódující tvorbu transkripčního faktoru, který hraje významnou úlohu v růstu dlouhých kostí. SHOX gen nepodléhá X-inaktivaci a za fyziologických okolností je přítomen ve dvou funkčních kopiích. Haploinsuficience SHOX genu (v důsledku delece nebo bodové mutace) se vyskytuje u 2,4 % jedinců s malým vzrůstem (< -2,0 SDS) a bývá provázena kostními odchylkami: Madelungovou deformitou předloktí, zkrácením středních segmentů dlouhých kostí (mezomelie), výraznějším zakřivením radia a tibie, kratšími metakarpy a metatarzy (především krátký IV. metakarp nebo metatarz) a gotickým patrem. Insuficience SHOX genu zahrnuje kontinuum klinických jednotek s různou tíží projevů: od závažného postižení u pacientů s Langerovým syndromem (homozygotní formou defektu SHOX genu), přes mírnější fenotyp u Lériho-Weillova syndromu (LWS) až k izolované růstové retardaci (ISS). Delece SHOX je rovněž součástí Turnerova syndromu; ten také pomohl v roce 1997 gen identifikovat a stal se výchozím modelem pro léčbu růstovým hormonem. Podávání růstového hormonu pacientům s haploinsuficiencí SHOX genu významně ovlivňuje růstovou rychlost a tím zlepšuje prognózu výšky v dospělosti. Nálezy jsou srovnatelné s výsledky léčby somatotropním hormonem u dívek s Turnerovým syndromem. Prokázaný deficit SHOX genu (potvrzený DNA analýzou) se stal v roce 2008 jednou z indikací k léčbě růstovým hormonem i v České republice.
Úvod
Tělesný růst představuje komplexní biologický fenomén, který je ovlivňován řadou vnitřních a vnějších faktorů. Incidence malého vzrůstu je v pediatrické populaci 3 : 100. Hormonální porucha jako příčina malé postavy se vyskytuje pouze u 1–2 % dětí, růstová retardace častěji provází chronická onemocnění systémové povahy (15 %), u nichž vzniká na základě různých patofyziologických mechanismů spojených se základním onemocněním (malnutrice, chronický zánět, hypoxie, acidóza a další). Růstová retardace je častým průvodním jevem i geneticky podmíněných syndromů (Downův, Turnerův, Noonanové syndrom) a kostních dysplazií, které bývají považovány za raritní. Disproporcionalita, jež tyto syndromy od raného věku většinou provází, je nepřehlédnutelná, proto bývají záhy diagnostikovány. Skeletálních dysplazií je dosud známo více než 350 typů a ne u všech byla identifikována jejich genová podstata. Většina dětí s malým vzrůstem (80 %) je malých, ale zdravých. Děti trpí buď tzv. konstitučním opožděním růstu (a často i puberty), nebo tzv. familiárně podmíněnou malou postavou; ta může být způsobena nenápadnou kostní dysplazií, která nemusí být vždy spojena s tělesnou disproporcionalitou.
SHOX gen
Při sledování klinických příznaků u početných souborů pacientek s Turnerovým syndromem bylo zjištěno, že většina genů, které mají význam v patogenezi somatických odchylek u Turnerova syndromu, je lokalizována do tzv. hlavní pseudoautozomální oblasti (PAR1) na distální části chromozomu X. Deficit celé pseudoautozomální oblasti nebo její části (haploinsuficience) vede ke ztrátě genové exprese.
Geny v tzv. pseudoautozomální oblasti (PAR) chromozomu X, která se nachází v distální části krátkých i dlouhých ramen chromozomu X, mají své homologní geny ve stejné lokalizaci na chromozomu Y a nepodléhají X-inaktivaci. Hlavní pseudoautozomální oblast (PAR1) je lokalizována na konci krátkého raménka chromozomu X i Y (Xp/Yp). V prvním meiotickém dělení dochází ke vzájemné výměně genů lokalizovaných v PAR mechanismem označeným jako crossing-over. Takto může dojít k přenosu genu z původní lokalizace na chromozomu X na chromozom Y, a tedy k tzv. X-linked přenosu onemocnění z otce na syna. Dědičný přenos připomíná autozomální dominanci. Geny, které se nacházejí v pseudoautozomální oblasti obou chromozomů X u žen a v pseudoautozomální oblasti chromozomu X a Y u mužů, jsou za fyziologických okolností přítomny ve dvou funkčních kopiích.
V roce 1997 byl v PAR1 obou pohlavních chromozomů (Xp22.3/Yp11.3) lokalizován homeobox obsahující gen, který byl nazván SHOX (Short stature HOmeoboX-containing gene on the X chromosome) [1]. SHOX gen nepodléhá X-inaktivaci a je za fyziologických okolností přítomen u obou pohlaví ve dvou funkčních kopiích. SHOX představuje regulační gen kódující tvorbu jednoho z proteinů s pravděpodobnou funkcí transkripčního faktoru. Bylo prokázáno, že k jeho expresi dochází u lidských embryí nejdříve v ektodermálních základech končetin kolem 32. dne intrauterinního života. Při 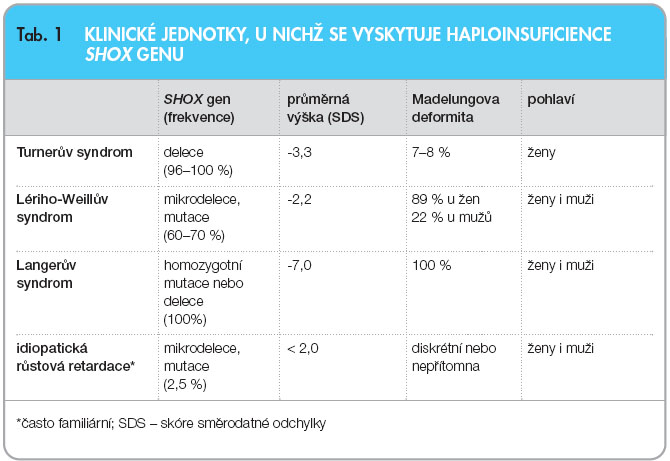 kondenzaci mezenchymu v prekartilaginózní primordiální kosti je exprese SHOX genu soustředěna do střední a distální části vyvíjejících se horních i dolních končetin (distální části humeru, radia a ulny a některých zápěstních kůstek, distální části femuru, tibie a fibuly a metatarzálních kůstek). Exprese SHOX genu není omezena pouze na končetiny. SHOX gen se uplatňuje i při vývoji tkání odvozených od mezenchymálních základů prvních dvou žaberních oblouků (dolní a horní čelist a kostní struktury formující střední a zevní ucho). Jeho exprese nebyla nalezena při vývoji axiálního skeletu a lebky, ani při vývoji vnitřních orgánů. SHOX gen tedy hraje zásadní roli ve vývoji skeletu, zejména předloktí a bérců.
kondenzaci mezenchymu v prekartilaginózní primordiální kosti je exprese SHOX genu soustředěna do střední a distální části vyvíjejících se horních i dolních končetin (distální části humeru, radia a ulny a některých zápěstních kůstek, distální části femuru, tibie a fibuly a metatarzálních kůstek). Exprese SHOX genu není omezena pouze na končetiny. SHOX gen se uplatňuje i při vývoji tkání odvozených od mezenchymálních základů prvních dvou žaberních oblouků (dolní a horní čelist a kostní struktury formující střední a zevní ucho). Jeho exprese nebyla nalezena při vývoji axiálního skeletu a lebky, ani při vývoji vnitřních orgánů. SHOX gen tedy hraje zásadní roli ve vývoji skeletu, zejména předloktí a bérců.
Klinické projevy insuficience SHOX genu
![Obr. 1 Tělesná výška v závislosti na počtu SHOX genů: a) pacienti bez SHOX genu – Langerův syndrom, b) přítomen jen jeden SHOX gen – Turnerův a Lériho-Weillův syndrom, c) přítomnost dvou SHOX genů – fyziologický nález, d) pacientka se třemi SHOX geny – nadměrný vzrůst; podle [13] – Blum, et al., 2007.](https://www.remedia.cz/photo-a-28135---.jpg) Haploinsuficience SHOX genu není typická jen pro pacientky s Turnerovým syndromem, u nichž gen chybí [2], SHOX gen může být rovněž inaktivní v d
Haploinsuficience SHOX genu není typická jen pro pacientky s Turnerovým syndromem, u nichž gen chybí [2], SHOX gen může být rovněž inaktivní v d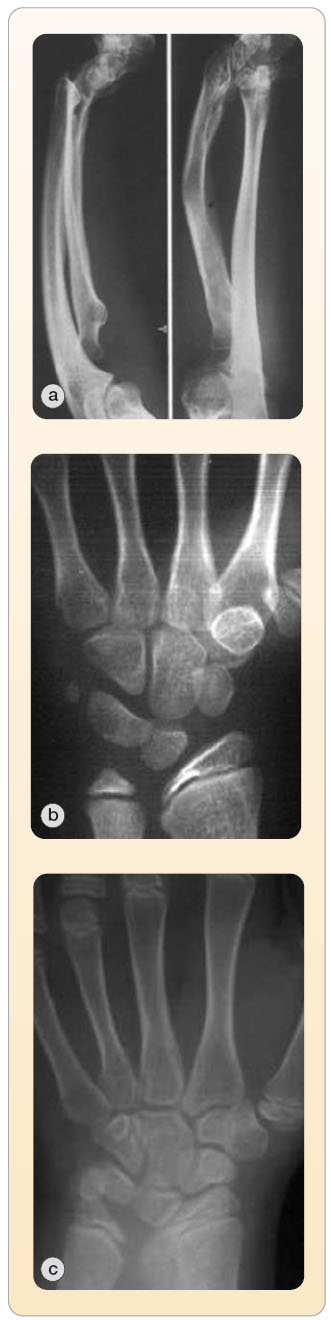 ůsledku bodové mutace nebo delece postihující obě pohlaví. Mezi pacienty s deficitem SHOX genu existuje velká fenotypická různorodost (obr. 1, tab. 1). Klasickým příznakem je variabilně disproporcionální růstová porucha mezomelického typu. Výška může v jednotlivých případech kolísat od těžké růstové retardace až po dolní hranici normálních hodnot [3, 4]. Značná rozdílnost je pozorována také ve výskytu kostních odchylek, které jsou častější i nápadnější u dívek a jejichž závažnost stoupá s přibývajícím věkem. Patří mezi ně především Madelungova deformita předloktí: zakřivení a zkrácení radia s dorzální subluxací hypoplastické distální části ulny, jež má za následek vklínění a deformaci karpálních kůstek a obvykle také bolestivou a omezenou hybnost zápěstí (obr. 2). Především u dívek dochází často k předčasnému uzávěru mediální části distální epifýzy ulny, který deformitu ještě zhoršuje.
ůsledku bodové mutace nebo delece postihující obě pohlaví. Mezi pacienty s deficitem SHOX genu existuje velká fenotypická různorodost (obr. 1, tab. 1). Klasickým příznakem je variabilně disproporcionální růstová porucha mezomelického typu. Výška může v jednotlivých případech kolísat od těžké růstové retardace až po dolní hranici normálních hodnot [3, 4]. Značná rozdílnost je pozorována také ve výskytu kostních odchylek, které jsou častější i nápadnější u dívek a jejichž závažnost stoupá s přibývajícím věkem. Patří mezi ně především Madelungova deformita předloktí: zakřivení a zkrácení radia s dorzální subluxací hypoplastické distální části ulny, jež má za následek vklínění a deformaci karpálních kůstek a obvykle také bolestivou a omezenou hybnost zápěstí (obr. 2). Především u dívek dochází často k předčasnému uzávěru mediální části distální epifýzy ulny, který deformitu ještě zhoršuje.
-
zkrácení středních segmentů dlouhých kostí (mezomelie),
-
výraznější zakřivení radia a tibie,
-
exostózy na proximálních částech tibie a fibuly,
-
kratší metakarpy a metatarzy (především čtvrtý),
-
gotické patro,
-
nápadná svalová hypertrofie, především na dolních končetinách.
Turnerův syndrom
Lériho-Weillův syndrom (dyschondroosteosis)
Kombinace výskytu Madelungovy deformity předloktí se zkrácením a zakřivením bérců a se závažnou růstovou poruchou je charakteristická pro osoby s familiárně dědičným Lériho-Weillovým syndromem, který byl poprvé popsán v roce 1929. Jeho incidence je 1 : 5000. Dědí se pseudoautozomálně dominantně. Postihuje tedy obě pohlaví, ale nápadnější je u žen, u nichž je v průměru čtyřikrát častěji diagnostikován než u mužů. Příčinou inaktivity SHOX genu u pacientů s Lériho-Weillovým syndromem je genová mutace (19 %) nebo delece (81 %) [7]. Výškový deficit kolísá od -6,4 do -0,6 SDS. Na rozdíl od dívek s Turnerovým syndromem je Madelungova deformita u pacientů s dyschondroosteózou dominantním příznakem, dále se typicky vyskytuje výrazná svalová hypertrofie postihující zejména dolní končetiny.
Langerův syndrom (mezomelická kostní dysplazie)
Extrémní případ inaktivace obou SHOX genů (pacienti jsou v podstatě homozygotní formou Lériho-Weillovy dyschondroosteózy) vede ke vzniku sporadicky se vyskytující mezomelické kostní dysplazie. Poprvé se o této chorobě zmiňuje radiolog Brailsford v roce 1935. (Podrobně byla charakterizována až o více než 30 let později Langerem, který popisuje případy dvou dětí s „mezomelickým trpaslictvím, hypoplastickou ulnou, fibulou a mandibulou“. Jejich rodiče byli bratranec a sestřenice a oba byli menší postavy, měli krátké prsty a deformovaná zápěstí). Průměrná výška pacientů s Langerovým syndromem je významně redukována (-7 SDS), především v důsledku výrazného zkrácení končetin, které je nápadné již od raného věku. Kosti bývají často deformovány a postiženy patologickými srůsty. Konstantním příznakem je Madelungova deformita, hypoplazie mandibuly a výrazná lordóza páteře [8].
Idiopatická růstová retardace
Děti s tzv. idiopatickou růstovou retardací (ISS) tvoří heterogenní skupinu jedinců, u kterých nebyla příčina malého vzrůstu dostupnými metodami zjištěna. Velmi dlouhou dobu byly považovány za tzv. variantu normy neboli děti „malé, ale zdravé“ a empiricky se dělily na děti s konstitučním opožděním růstu a děti s familiární růstovou retardací.
Výsledky četných studií prokázaly defekt SHOX genu přibližně u 2–3 % dětí s idiopatickou růstovou retardací (obvykle familiárního typu), u nichž nebyly v době diagnózy patrny žádné kostní změny nebo byly jen velmi nenápadně vyjádřeny (nejčastěji se vyskytuje krátký IV. metakarp!). Příčinou bývá mikrodelece SHOX genu, vzácněji jeho bodová mutace [9, 10].
Nadbytečný počet SHOX genů mají jedinci s nadpočetným chromozomem X nebo Y (např. 47,XXX, 47,XXY nebo 47,XYY) nebo osoby s parciální duplikací chromozomu X. Jsou vyšší postavy, mají delší dolní segment, větší rozpětí paží. Dle některých autorů mají prodlouženou periodu tělesného růstu [11].
Diagnostika insuficience SHOX genu
 Na insuficienci SHOX genu by mělo být pomýšleno u všech dětí postižených familiární poruchou růstu nejasné etiologie a u pacientů s výše popsanými kostními odchylkami. K racionální indikaci k vyšetření SHOX genu nám může pomoci jednoduchý skórovací systém, který je uveden v tab. 2. Při podezření na insuficienci SHOX je nutné odeslat dítě k dalšímu vyšetření do specializované ordinace pro poruchy růstu, případně indikovat přímo genetické vyšetření. Diagnostika delece nebo mutace SHOX genu se v současné době provádí i v České republice.
Na insuficienci SHOX genu by mělo být pomýšleno u všech dětí postižených familiární poruchou růstu nejasné etiologie a u pacientů s výše popsanými kostními odchylkami. K racionální indikaci k vyšetření SHOX genu nám může pomoci jednoduchý skórovací systém, který je uveden v tab. 2. Při podezření na insuficienci SHOX je nutné odeslat dítě k dalšímu vyšetření do specializované ordinace pro poruchy růstu, případně indikovat přímo genetické vyšetření. Diagnostika delece nebo mutace SHOX genu se v současné době provádí i v České republice.
Možnosti léčby
Od 80. let minulého století, s příchodem rekombinantního růstového hormonu (GH) na farmaceutický trh, se začaly hledat nové indikace k jeho použití. Do té doby byli léčeni (substituováni) pouze pacienti s deficitem růstového hormonu. Úspěšné klinické studie vedly k tomu, že se léčba postupně rozšířila i na dívky s Turnerovým syndromem (1992), na pacienty s malým vzrůstem trpící chronickou renální insuficiencí (1995), na dospělé pacienty s prokázaným těžkým deficitem GH (1998), na pacienty se syndromem Pradera-Williho, kteří mají nedostatek GH v důsledku neurosekretorické (hypotalamické) poruchy (2001), a na děti s postnatálním růstovým selháním navazujícím na intrauterinní růstovou retardaci (2003).
Právě Turnerův syndrom se stal výchozím modelem nejen pro diagnostiku insuficience SHOX, ale i pro její případnou terapii. Již více než dvacet let jsou pacientky s Turnerovým syndromem léčeny farmakologickými dávkami GH. Výsledky randomizovaných klinických studií u nich potvrdily nejen krátkodobé zlepšení růstové rychlosti, ale i velmi dobrý vliv na výšku v dospělosti [12]. V roce 2007 byly zveřejněny výsledky první multicentrické randomizované studie u pacientů s prokázaným defektem SHOX, která měla zhodnotit:
-
četnost defektu v SHOX genu mezi dětmi s růstovou retardací,
-
u potvrzených SHOX deficitů zhodnotit účinnost léčby GH a porovnat ji s účinkem léčby u dívek s Turnerovým syndromem.
 podávání GH dívkám s Turnerovým syndromem [13, 14] (tab. 3). Defekt SHOX genu se tak stal jednou z indikací k léčbě GH ve všech vyspělých zemích světa, od roku 2008 i v České republice. Podmínkou léčby je prokázání defektu SHOX. Protože z terapie mají přínos zejména ti pacienti, u kterých byla léčba zahájena v prepubertálním věku, je důležitá včasná diagnóza této kostní dysplazie. Může na ni upozornit zejména familiární výskyt malé postavy v několika po sobě jdoucích generacích a eventuální přítomnost popsaných kostních odchylek.
podávání GH dívkám s Turnerovým syndromem [13, 14] (tab. 3). Defekt SHOX genu se tak stal jednou z indikací k léčbě GH ve všech vyspělých zemích světa, od roku 2008 i v České republice. Podmínkou léčby je prokázání defektu SHOX. Protože z terapie mají přínos zejména ti pacienti, u kterých byla léčba zahájena v prepubertálním věku, je důležitá včasná diagnóza této kostní dysplazie. Může na ni upozornit zejména familiární výskyt malé postavy v několika po sobě jdoucích generacích a eventuální přítomnost popsaných kostních odchylek. Závěr
Včasné rozpoznání pacientů s defektem v SHOX genu je důležité z několika důvodů:
-
Vede k přesné diagnostice růstové poruchy.
-
Je možno je využít v rámci prekoncepčního nebo prenatálního poradenství u postižených jedinců.
-
Haploinsuficience SHOX může být součástí rozsáhlejšího genového defektu a u chlapců může vést k postupnému vývoji jiných závažných onemocnění, která je v případě potvrzení diagnózy možno předvídat (např. chondrodysplazie, mentální retardace nebo Kallmannův syndrom).
-
Pacienti s insuficiencí SHOX mohou mít významný přínos z léčby GH.
Práce byla podpořena projekty MSM 0021620820 a IGA MZ ČR NS/9743-4.
Seznam použité literatury
- [1] Rao E, Weiss B, Fukami M, et al. Pseudoautosomal deletions encopassing a novel homeobox gene cause growth failure in idiopathic short stature and Turner syndrome. Nat Genet 1997; 16: 54–63.
- [2] Clement-Jones M, Schiller S, Rao E, et al. The short stature homeobox gene SHOX is involved in skeletal abnormalities in Turner syndrome. Hum Mol Genet 2000; 9: 695–702.
- [3] Rappold GA, Fukami M, Niesler B, et al. Deletions of the homeobox gene SHOX (short stature homeobox) are an important cause of growth failure in children with short stature. J Clin Endocrinol Metab 2002; 87: 1402–1406.
- [4] Ross JL, Scott C Jr, Martilla P, et al. Phenotypes Associated with SHOX Deficiency. J Clin Endocrinol Metab 2001; 86: 5674–5680.
- [5] Jacobs P, Dalton P, James R, et al. Turner syndrome: a cytogenetic and molecular study. Ann Hum Genet 1997; 61: 471–483.
- [6] Ogata T, Muroya K, Matsuo A, et al. Turner syndrome and Xp deletions: clinical and molecular studies in 47 patients. J Clin Endocrinol Metab 2001; 86: 5498–5508.
- [7] Schiller S, Spranger S, Schechinger B, et al. Phenotypic variation and genetic heterogeneity in Léri-Weill syndrome. Eur J Hum Gen 2000; 8: 54–62.
- [8] Zinn AR, Wei F, Zhang L, et al. Complete SHOX deficiency causes Langer mesomelic dysplasia. Am J Med Genet 2002; 110: 158–163.
- [9] Rappold G, Blum WF, Shavrikova EP, et al. Genotypes and phenotypes in children with short stature: clinical indicators of SHOX haploinsufficiency. J Med Genet 2007; 44: 306–313.
- [10] Huber C, Rosilio M, Munnich A, et al. High incidence of SHOX anomalies in individuals with short stature. J Med Genet 2006; 43:735–739.
- [11] del Rey G, Jasper H, Bengolea SV, et al. Trisomy of the short stature homeobox-containing gene (SHOX) due to duplication/deletion of the X chromosome: clinical implications on the stature. Horm Res Paediatr 2010; 74: 297–304.
- [12] Stephure DK, Canadian Growth Hormone Advisory Commitee. Impact of growth hormone supplementation on adult height in Turner syndrome: results of the Canadian randomized controlled trial. J Clin Endocrinol Metab 2005; 90: 3360–3366.
- [13] Blum FW, Crowe BJ, Quigley CA, et al. Growth hormone is effective in treatment of short stature associated with short stature homeobox-containing gene deficiency: Two-year results of a randomized, controlled, multicenter trial. J Clin Endocrinol Metab 2007; 92: 219–228.
- [14] Price DA, Larsson P, Darendellier F, et al. Response to growth hormone treatment in Léri-Weill dyschondrosteosis (LWD): Analysis of data from KIGS. Horm Res 2002; 57: p. 56.