Farmakoterapie plicní arteriální hypertenze
Plicní arteriální hypertenze (PAH) je závažné a nevyléčitelné onemocnění charakterizované progredující prekapilární plicní hypertenzí. PAH postihuje především mladší ženy a neléčená vede během 2–3 let od diagnózy ke smrti. Vazodilatační léčba blokátory kalciových kanálů je vhodná jen u menšiny pacientů, kteří mají zachovalou vazoreaktivitu plicních cév. V posledních letech jsme svědky velkého pokroku v terapii PAH. V randomizovaných studiích byla zkoušena s úspěchem řada nových přípravků (prostanoidy, antagonisté endotelinových receptorů, inhibitory fosfodiesterázy 5), které zasahují na různé úrovni do patogenetických mechanismů PAH a zlepšují funkční zdatnost, hemodynamiku a prognózu nemocných. Kauzální terapii však nadále nemáme k dispozici. Limitem nových léčebných postupů zůstává řada nežádoucích účinků a ekonomická náročnost. Terapie navíc vyžaduje velmi individuální přístup ke každému pacientovi. Proto je nezbytné soustředit nemocné do specializovaných center, která jsou schopna poskytnout komplexní diagnostiku i léčbu.
Úvod a klasifikace
Plicní hypertenze je syndrom charakterizovaný zvýšením středního tlaku v plicnici nad 25 mmHg v klidu nebo nad 30 mmHg při zátěži [1].
Letité snahy podrobněji klasifikovat chronickou plicní hypertenzi vyústily v roce 2003 v tzv. Benátskou klasifikaci plicní hypertenze [2]. Klasifikace rozeznává 5 kategorií plicní hypertenze (tab. 1).
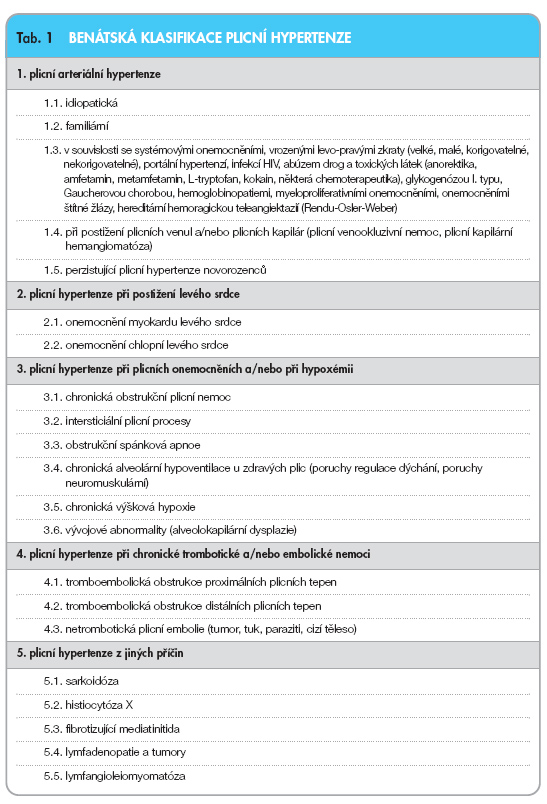
Klinické jednotky v každé kategorii mají do jisté míry podobnou patogenezi, histologický obraz, kliniku a léčbu. Klasifikace však pomíjí hledisko funkční a genetické. Jde tedy o nomenklaturu především klinickou, která dovoluje jednoznačnou komunikaci v oblasti plicní hypertenze, standardizaci diagnostiky a léčby, koncipování klinických studií a studium patofyziologických mechanismů v jasně definovaných populacích pacientů. Podle stupně závažnosti rozlišujeme plicní hypertenzi lehkou, středně těžkou a těžkou (tab. 2).

K vyjádření funkční zdatnosti u plicní hypertenze se používá modifikované klasifikace podle NYHA (tab. 3) [3].

Plicní arteriální hypertenze (PAH) představuje skupinu chorob, pro kterou je charakteristická přítomnost prekapilární plicní hypertenze, v histologickém obraze bývá nález plexiformní arteriopatie a v léčbě je typický efekt prostanoidů, antagonistů endotelinových receptorů a inhibitorů fosfodiesterázy [4].
Roční incidence idiopatické a familiární PAH v běžné západní populaci se odhaduje na 1–2 případy na 1 milion obyvatel. Průměrný věk nemocných je 35–40 let. Ženy jsou postiženy asi 1,7x častěji než muži. Přibližně 6 % případů je familiárního původu, ostatní se označují jako idiopatické [2]. V případě vaskulitid se s plicní hypertenzí setkáváme nejčastěji u systémové sklerodermie. Incidence PAH se pohybuje od 6 % do 60 %. V případě difuzní formy sklerodermie trpí plicní hypertenzí až 33 % nemocných. U CREST (kalcinóza, Raynaudův fenomén, ezofageální dysmotilita, sklerodaktylie, teleangiektazie) varianty systémové sklerodermie se plicní hypertenze vyskytuje ve více než 60 % případů. V případě systémového lupus erythematodes je plicní hypertenze přítomna u 4–14 % pacientů. Asi u 20 % pacientů s revmatoidní artritidou, kteří nemají současně jiné plicní nebo srdeční onemocnění, lze zjistit lehkou plicní hypertenzi. Plicní hypertenze u nemocných s portální hypertenzí se vyskytuje asi ve 2 % případů [5]. U pacientů infikovaných virem HIV je incidence plicní hypertenze zhruba 0,5 % [6]. Zvýšení incidence plicní hypertenze u nemocných užívajících anorektika bylo poprvé popsáno v 60. letech dvacátého století po zavedení aminorex fumarátu. O 20 let později bylo zaznamenáno zvýšení incidence plicní hypertenze u nemocných léčených fenfluraminem a dexfenfluraminem. K rozvoji plicní hypertenze vede většinou delší než šestiměsíční užívání přípravku [7].
Patofyziologie
Klinické jednotky řazené do skupiny PAH jsou značně heterogenní, určité podstatné rysy v patofyziologii onemocnění jsou však společné.
Rozvoj PAH je pravděpodobně dán kombinací faktorů genetických a zevních [8]. Asi u 60 % případů familiární PAH, u 25 % případů idiopatické PAH a u 10 % nemocných s PAH vzniklou v souvislosti s užíváním anorektik nalézáme mutaci genu pro receptor BMPR II (bone morphogenetic protein). Dědičnost je autosomálně dominantní s neúplnou penetrancí. Gen BMPR2 je lokalizován na dlouhém raménku 2. chromozomu (2q31–32) [9]. U nemocných s PAH nalézáme řadu dalších abnormit. Dysfunkční endotel produkuje ve zvýšené míře endotelin, který má výrazné vazokonstrikční a mitogenní vlastnosti, zatímco syntéza vazodilatačního prostacyklinu a NO je snížena. Buňky hladkého svalstva zvýšeně exprimují serotoninové transportéry, a tak umožňují vazokonstrikční a mitogenní působení cirkulujícího serotoninu. Dysfunkční voltážově řízené draslíkové kanály rovněž usnadňují proliferaci. V adventicii plicních cév je zvýšeně produkován silně mitogenní tenascin. Dochází k aktivaci trombocytů, ztrátě tromborezistence endotelu a k aktivaci koagulační kaskády. Důsledkem všech těchto dějů je remodelace plicních cév, vznik plexiformních lézí a trombózy in situ [10].
Vyvolávající faktor u idiopatické a familiární PAH není znám. V ostatních případech PAH je tímto faktorem vaskulitida, hyperkinetická cirkulace, virová infekce (virus HIV, ale také zřejmě herpes virus HHV-8 a virus hepatitidy C), nebo abúzus anorektik.
Morfologické změny na plicních cévách u PAH mají charakter proliferativní a obliterativní. Jsou progresivní a postupně vedou k obstrukci a restrikci plicního cévního řečiště. Důsledkem je progrese plicní hypertenze. Při zvyšujícím se mechanickém namáhání cévní stěny dochází k dalšímu zhoršování endoteliální dysfunkce.
Přestože žádná z výše popsaných abnormit nemůže sama o sobě vysvětlit patofyziologii PAH, poznání dílčích mechanismů umožňuje alespoň paliativní terapeutickou intervenci. Dokladem toho je např. úspěšná léčba analogy prostacyklinu, antagonisty endotelinových receptorů nebo inhibitory fosfodiesterázy.
Prognóza
Prognóza neléčené PAH není závislá na věku, pohlaví a délce trvání symptomů v době stanovení diagnózy. Závisí na výši tlaku v plicnici a na plicním cévním odporu, na vazodilatační odpovědi plicních cév, na přítomnosti pravostranného srdečního selhání, saturaci smíšené žilní krve kyslíkem a na funkční zdatnosti. Nejspolehlivějším prediktivním ukazatelem je pravděpodobně střední tlak v pravé síni.
K prognosticky nepříznivým známkám patří dilatace pravé síně a přítomnost perikardiálního výpotku, vzdálenost dosažená při testu šestiminutové chůze pod 330 m, pokles saturace při testu šestiminutové chůze o více než 10 %, trvající funkční stadium NYHA III-IV i při specifické léčbě, hladina BNP nad 150 pg/ml, vyšší hladina ANP, norepinefrinu, endotelinu-1, kyseliny močové a pozitivní troponin T.
Jako průměrná doba přežití od stanovení diagnózy se uvádí 2,8 roku. Průměrné přežití ve stadiu NYHA IV je 6 měsíců, ve stadiu NYHA III 2,5 roku, ve stadiu NYHA I a II asi 5 let. Očekávané přežití u neléčených pacientů je 1 rok 68 %, 3 roky 48 % a 5 let 34 % [11, 12].
Vývoj a principy farmakoterapie
Cílem farmakoterapie PAH je zasáhnout do 3 základních patofyziologických mechanismů uplatňujících se při vzniku a rozvoji plicní arteriální hypertenze (vazokonstrikce, proliferace, prokoagulační stav) a ovlivnit symptomy a prognózu nemocných.
Vývoj léčebných postupů PAH v posledních padesáti letech byl značně pomalý (tab. 4).

První pokusy ovlivnit onemocnění se datují do 50. let dvacátého století, kdy se empiricky zkoušelo podání řady látek s vazodilatačním účinkem s cílem ovlivnit tlak v plicnici. V 70. a 80. letech minulého století bylo provedeno několik malých nekontrolovaných studií s dlouhodobým podáním vazodilatancií (sublingvální izoprenalin, dále diazoxid, hydralazin). Cílem studií bylo ověřit hypotézu, že vazokonstrikce je hlavní patogenetický mechanismus u PAH. Výsledky byly povzbudivé zejména v ovlivnění funkční zdatnosti a prognózy. Během dalších 10 let byly publikovány práce o efektu antikoagulační léčby a terapii vysokými dávkami blokátorů kalciových kanálů. Tento léčebný postup se spolu s podáním diuretik, digoxinu a kyslíku označuje a stále používá jako konvenční léčba PAH.
V 90. letech byl v několika studiích dokumentován příznivý vliv kontinuální intravenózní infuzní léčby epoprostenolem. Tato terapie se označuje jako nekonvenční pro komplikovaný způsob podání, který zahrnuje implantaci tunelizovaného katétru a kontinuální dávkování léku malou přenosnou pumpou [13].
V posledních letech byla provedena řada kontrolovaných zaslepených studií s tzv. novými přípravky (analoga prostacyklinu, antagonisté endotelinových receptorů, inhibitory fosfodiesterázy) (obr. 1).
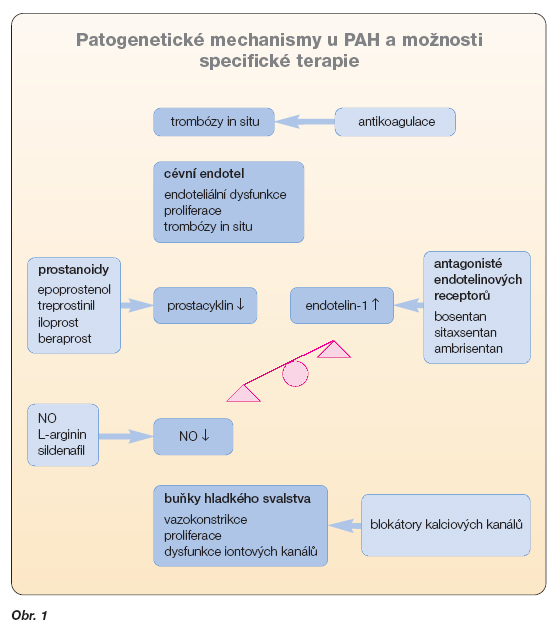
Studie sledovaly většinou tříměsíční účinek léčby. Výsledky lze považovat za hodnotitelné, neboť progrese symptomatického onemocnění u neléčeného pacienta je vždy zřetelná v intervalu kratším než 3 měsíce. Velikost sledovaných souborů je omezena vzácným výskytem onemocnění. Jako primární cíl je většinou hodnocena tolerance zátěže měřená testem šestiminutové chůze. Naopak změna hemodynamických parametrů bývá sledována jako cíl sekundární, neboť rutinně měřené klidové hodnoty mají omezenou výpovědní hodnotu a pravděpodobně na rozdíl od hodnot stanovených při zátěži jen velmi málo korelují se změnou tolerance fyzické zátěže. Vzájemné srovnání randomizovaných studií přináší některé svízele. Problémem je nehomogenní zastoupení jednotlivých typů PAH ve studovaných populacích a dále nejednotná, a tudíž obtížně srovnatelná definice stupňů podle klasifikace NYHA, zejména NYHA IV. Studie nebyly navrženy jako mortalitní. Nadále proto zůstává nutnost srovnání s historickými kontrolami. Bezpečnost jednotlivých léků a jejich účinek prokázaný v randomizovaných studiích je průběžně studován v otevřených studiích [14].
Na základě závažnosti poznatků z klinických studií bývají léčebné postupy klasifikovány stupni A–C (tab. 5) [15].

Přes veškerý pokrok ve farmakoterapii má léčba PAH stále do jisté míry charakter paliativní terapie, ač některé léčebné postupy prodlužují dobu přežívání nemocných. Léčba každého pacienta s plicní arteriální hypertenzí může být provázena nepředpokládanými vedlejšími účinky, proto vyžaduje velkou obezřetnost při indikaci, zahájení i vedení.
Konvenční léčba
Blokátory kalciových kanálů
Blokátory kalciových kanálů vedou ke zlepšení hemodynamiky a funkce pravé komory. Prognózu zlepšují u pacientů s tlakem v pravé síni nižším než 10 mmHg. Léčba je indikována pouze u nemocných s pozitivním testem akutní plicní vazodilatace. Principem testu je sledování změn hemodynamických parametrů po podání dobře titrovatelného vazodilatancia, které je alespoň relativně selektivní pro plicní cirkulaci (inhalace NO, intravenózní prostacyklin nebo intravenózní adenosin) (tab. 6).

Retrospektivní analýzou rozsáhlého souboru 557 pacientů s PAH bylo prokázáno, že ač test byl pozitivní asi ve 13 % případů, dlouhodobě profitovalo z léčby blokátory kalciových kanálů pouze 7 % nemocných [16]. Doporučují se vysoké, maximálně tolerované dávky (tab. 7) [17].
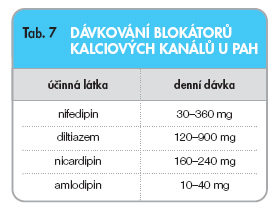
Terapie blokátory kalciových kanálů není vhodná u nemocných s porušenou funkcí pravé komory, se srdečním indexem pod 2,1 l/min./m2 a se saturací smíšené žilní krve kyslíkem pod 63 %, dále u nemocných s plicní venookluzivní nemocí a plicní kapilární hemangiomatózou.
Perorální antikoagulancia
Indikace antikoagulační léčby u PAH vychází z tradičně dokumentovaného prokoagulačného stavu a nálezu trombóz in situ u těchto nemocných [18]. Důkazy o účinku perorální antikoagulační léčby vycházejí z několika nerandomizovaných retrospektivních studií, které zahrnuly pouze nemocné s idiopatickou PAH a pacienty s PAH asociovanou s užíváním anorektik. V úvahách o indikaci antikoagulační léčby u ostatních typů PAH lze vycházet z těchto retrospektivních analýz při pečlivém zvážení poměru rizika a benefitu (vyšší riziko krvácení do trávicího traktu u nemocných s PAH při jaterních onemocněních, naopak vyšší riziko trombózy katétru u nemocných léčených epoprostenolem). V recentně provedených studiích léků PAH užívalo antikoagulační léčbu 51–86 % nemocných, zejména s diagnózou idiopatické PAH ve funkčním stadiu NYHA III a IV.
Cílové INR při léčbě antagonisty vitaminu K se má pohybovat kolem 2. U PAH v souvislosti se systémovými onemocněními pojiva při pozitivitě antifosfolipidových protilátek se doporučuje cílové INR 3.
Diuretika
Diuretika zlepšují symptomy v souvislosti s městnáním při srdečním selhání, nevedou však pravděpodobně k žádnému zlepšení prognózy, protože nezasahují do vlastních patogenetických mechanismů. V nedávno uskutečněných randomizovaných studiích u PAH užívalo diuretika 49–70 % zařazených jedinců.
V dávkování diuretik u nemocných s PAH je třeba velké opatrnosti, neboť snižují předtížení pravé komory jako zásadní parametr určující srdeční výdej. Diuretika také zvyšují viskozitu krve, a tak potencují riziko trombotických komplikací.
Kličková diuretika je vhodné dávkovat několikrát denně, jednorázová dávka nemá překročit 40–60 mg furosemidu. Denní negativní bilance by neměla být vyšší než odpovídající poklesu hmotnosti o 1 kg. Nevede-li k úspěchu konvenční dávka kličkového diuretika (do 160 mg furosemidu denně), je vhodná kombinace s thiazidovými a kalium šetřícími diuretiky [19].
Digoxin, dopamin, dobutamin
Intravenózní podání digoxinu u nemocných s PAH vede ke krátkodobému zlepšení srdečního výdeje a snížení hladiny cirkulujícího norepinefrinu. Data o dlouhodobějším účinku však scházejí. V případě současné systolické dysfunkce levé komory a tachyfibrilace síní zlepší digoxin systolickou funkci pravé komory. U ostatních nemocných by neměl být součástí léčebného schématu [20]. Intravenózní dopamin nebo dobutamin může akutně zlepšit symptomy pravostranného srdečního selhání, vliv na prognózu onemocnění není znám.
Oxygenoterapie
Většina případů PAH (s výjimkou PAH při vrozených levopravých zkratech) se v klidu manifestuje jen výjimečně výraznou hypoxémií. V současné době neexistují jednoznačné důkazy o příznivém vlivu dlouhodobé oxygenoterapie na plicní hemodynamiku [20]. Efekt oxygenoterapie je však zejména ve zlepšení oxygenace tkání. Velmi kontroverzní je indikace oxygenoterapie u pacientů s Eisenmengerovým syndromem. Některé práce dokládají nedostatečný účinek noční oxygenoterapie na změnu hematologických parametrů, kvalitu života a přežívání. Údaje o účinku dlouhodobé domácí oxygenoterapie (15–16 hodin denně) postrádáme.
Prostanoidy
Prostacyklin (prostaglandin I2) je hlavní produkt metabolismu kyseliny arachidonové v cévách. Jeho účinky zahrnují zejména vazodilataci v plicním i systémovém oběhu a inhibici agregace a adheze trombocytů cestou stimulace adenylátcyklázy, která vede ke zvýšení cyklického adenosinmonofosfátu (cAMP) v krevních destičkách. Je pravděpodobné, že prostacyklin působí antiproliferačně a brání cévní remodelaci. Farmakokinetika prostacyklinu vysvětluje obtíže, s nimiž je spojeno jeho klinické užití. V organismu podléhá rychlé hydrolýze a enzymatické degradaci, biologický poločas je 2–3 minuty.
Pro PAH je charakteristická snížená sekrece prostacyklinu a nižší aktivita prostacyklinsyntázy v plicích [21].
Epoprostenol
Epoprostenol je syntetický analog prostacyklinu, který je v léčbě PAH používán již více než dvě desetiletí. Vzhledem ke krátkému biologickému poločasu je nutno jej podávat formou dlouhodobé kontinuální infuze do centrálního žilního katétru (obr. 2).
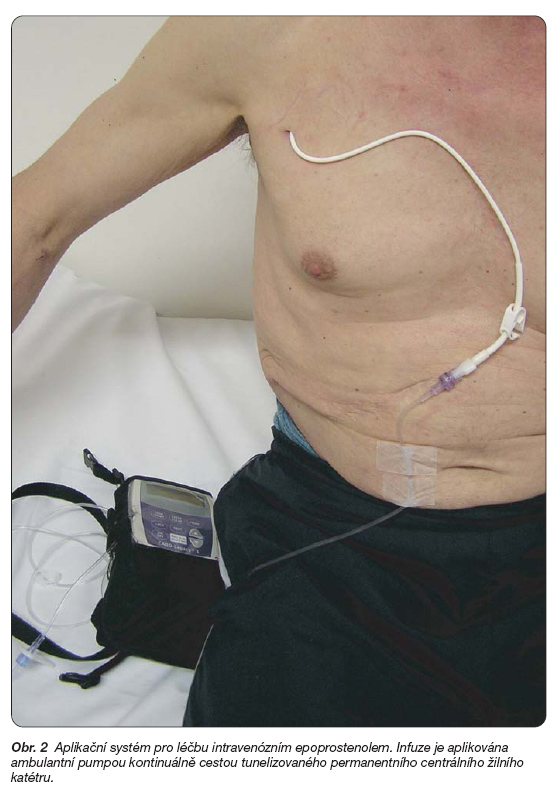
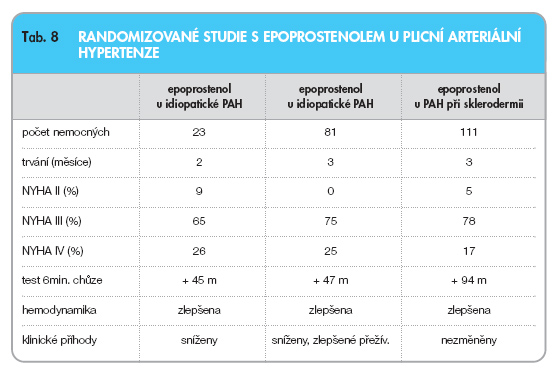
Vazodilatační účinky epoprostenolu na plicní cirkulaci u pacientů v pokročilém stadiu PAH nejsou podstatné. Důležité je především působení antiproliferační, antiagregační a pozitivně inotropní. Dlouhodobá kontinuální infuzní léčba epoprostenolem je indikována u nemocných s PAH ve III. a IV. stadiu funkční klasifikace NYHA, kteří nejsou respondéry na základě testu akutní plicní vazodilatace, případně u nich došlo k selhání vazodilatační léčby blokátory kalciových kanálů. Epoprostenol zlepšuje u těchto pacientů funkční zdatnost, hemodynamické parametry a kvalitu života. Je dokumentováno zlepšení prognózy s tříletým přežíváním v 65 % případů [22]. Výsledky randomizovaných studií s epoprostenolem jsou shrnuty v tab. 8 [23, 24].
Iniciální dávka epoprostenolu pro dlouhodobou infuzní léčbu se určí titrací přípravku. Dlouhodobá infuze do centrálního žilního katétru se pak zahajuje dávkou, která je o 4 ng/kg/min. nižší než maximální tolerovaná dávka. Pokud je maximální tolerovaná dávka nižší než 5 ng/kg/min., pak je počáteční terapeutická dávka polovinou maximální tolerované dávky.
Epoprostenol se dávkuje přenosnou pumpou, která musí splňovat tyto parametry: malá, lehká, dávkování preparátu po stupních 2 ng/kg/min. s přesností Î 6 % aplikované dávky, alarm přerušení infuze a slabých baterií. Náhlé přerušení infuze při poruše pumpy může vést k prudkému zhoršení stavu nemocného v důsledku rebound fenoménu. Proto se doporučuje, aby pacient měl k dispozici rezervní přístroj.
Epoprostenol je po rozpuštění relativně nestabilní. Nesmí být podáván v jedné infuzi současně s jinými preparáty. Roztok musí být chráněn před světlem a skladován při teplotě 2–8 °C. Při pokojové teplotě zůstává roztok stabilní 8 hodin, při chlazení sáčky s ledem 24 hodin.
V důsledku tachyfylaxe je nutno dávku epoprostenolu během chronické léčby zvyšovat. Průměrná dávka na začátku léčby činí 4–8 ng/kg/min., po roce léčby asi 20 ng/kg/min. U řady nemocných dojde při dávce 20–40 ng/kg/min. ke stabilizaci klinického stavu a kontinuální zvyšování dávek pak není nutné.
Vedle vlastních nežádoucích účinků epoprostenolu (bolesti čelistí, flush, bolesti hlavy, nauzea, hypotenze, tachykardie, bolesti na hrudi, trombocytopenie) jsou hlavním rizikem léčby lokální a systémové infekční komplikace v důsledku permanentního centrálního žilního katétru, a dále riziko poruchy infuzní pumpy. Ze zkušeností na velkých souborech nemocných lze očekávat lokální infekční komplikaci v oblasti centrálního žilního katétru s pravděpodobností 22–68 % ročně u každého nemocného a pravděpodobnost sepse 0–39 %. Porucha pumpy nebo problémy s průchodností katétru se vyskytují v těchto souborech 2–3krát během roku u každého pacienta [14].
Léčba epoprostenolem je mimořádně náročná na úzkou spolupráci mezi nemocným, jeho rodinou a centrem, které řídí léčbu. Pacient a jeho rodina musí být vyškoleni v ošetřování katétru, v přípravě léku a obsluze pumpy. Nezbytná je možnost telefonické konzultace s centrem, a to kdykoli. Pacient také telefonicky hlásí pravidelně v intervalu 1–2 týdny do centra svůj aktuální klinický stav, rychlost infuze, aktuální hodnotu INR a případně další údaje. Nejdelší dokumentované přežití nemocného léčeného infuzí epoprostenolu je 14 let.
Původně se léčba epoprostenolem považovala za most k transplantaci plic. Dnes jde o terapii první volby a o nejúčinnější alternativu transplantace u pacientů s těžkou PAH. Léčba epoprostenolem oddaluje nutnost transplantace až o 17 měsíců. Dále je dokumentováno zlepšení přežívání po transplantaci u nemocných, kteří byli před výkonem léčeni epoprostenolem. Léčba epoprostenolem snižuje riziko měsíční mortality o 66 %, zatímco transplantace pouze o 18 %.
Náklady na roční léčbu 1 pacienta představují průměrně 2,5 milionu Kč.
Treprostinil
Treprostinil je tricyklický benzidinový analog prostacyklinu, který je natolik stabilní, že může být podán rozpuštěný ve fyziologickém roztoku za pokojové teploty. Je možná intravenózní a subkutánní aplikace, zkouší se také podání inhalační. Subkutánní infuze je ve srovnání s intravenózní infuzí méně náročná na vybavení pumpou, a zejména je prosta komplikací spojených s implantovaným centrálním žilním katétrem (obr. 3).
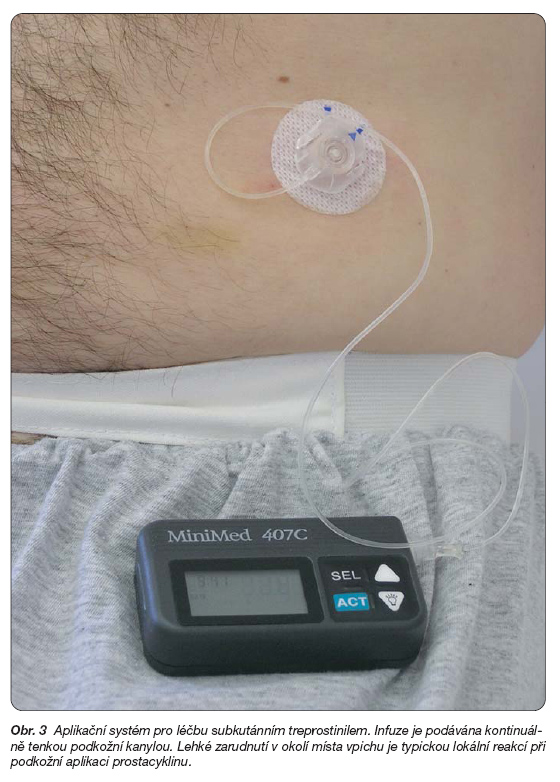
Při intravenózním podání odpadá ve srovnání s epoprostenolem nutnost chlazení infuzní soustavy, a zejména riziko plynoucí z náhlého přerušení infuze. Poločas léku při intravenózním podání je 27 minut, při subkutánním podání 58–83 minut.
Účinek léčby treprostinilem u pacientů s plicní arteriální hypertenzí byl studován v několika menších studiích, a především v rozsáhlé multicentrické studii, která zahrnula celkem 469 nemocných (tab. 9).
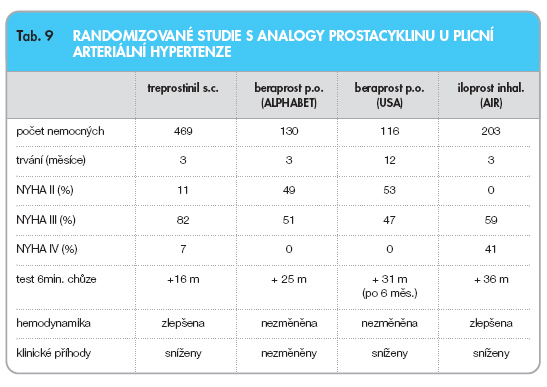
Ve skupině léčených došlo ke zlepšení tolerance zátěže a ke zlepšení hemodynamických parametrů ve srovnání s placebem. Nejvíce profitovali pacienti tolerující dávku vyšší než 13,8 ng/kg/min. (zlepšení při testu šestiminutové chůze o 36 m). Zlepšení bylo zřetelnější u nemocných ve stadiu NYHA III, IV než ve stadiu NYHA II [25, 26]. Je doložen rovněž vliv treprostinilu na zlepšení prognózy. Tříleté přežití je 69,8 % proti očekávaným 46 %.
Nejčastějšími nežádoucími účinky při podání treprostinilu je bolest v místě infuze, lokální reakce a zvracení. Bolest v místě subkutánní aplikace je na dávce nezávislá a individuálně vnímaná.
Jako iniciální dávka se doporučuje 1,25 ng/kg/min., u nemocných s hepatopatií 0,625 ng/kg/min. Počáteční terapeutická dávka má být nejméně 10 ng/kg/min. Má být dosažena postupným zvyšováním dávky každých 7 dní. Dále se titrace provádí podle dosaženého optimálního klinického efektu a minimálních nežádoucích účinků.
Indikace treprostinilu je obdobná jako u epoprostenolu (byl schválen pro léčbu pacientů s PAH ve stadiu NYHA II, III a IV). Jsou známy případy úspěšné konverze léčby z intravenózního epoprostenolu na subkutánní treprostinil. Ekonomická náročnost léčby je rovněž podobná jako u epoprostenolu.
Beraprost
Beraprost je perorální stabilní analog prostacyklinu. Stability je dosaženo díky cyklopentabenzofuranylovému skeletu. Preparát je ve střevě rychle resorbován. Účinky na hemodynamiku se začínají projevovat za 15–30 minut. Vrcholové plazmatické koncentrace dosahuje průměrně za 1,4 hodiny.
Do evropské multicentrické, prospektivní a placebem kontrolované studie AL-PHABET (Arterial Pulmonary Hypertension and Beraprost European Trial) bylo randomizováno celkem 130 pacientů s plicní arteriální hypertenzí (tab. 9).
Průměrná dávka beraprostu činila 80 mg 4x denně. Ve skupině léčené beraprostem došlo po 12 týdnech ke zlepšení tolerance zátěže hodnocené testem šetiminutové chůze, signifikantně však pouze v podskupině nemocných s idiopatickou PAH. Hemodynamické parametry se významně nezměnily [27]. Obdobné výsledky přinesla studie prováděná v USA. Tato studie navíc sice prokázala zlepšení vzdálenosti dosažené při testu šestiminutové chůze po 3 a 6 měsících léčby, efekt však již nebyl zřetelný po 9 a 12 měsících léčby. Beraprost rovněž vedl relativně často k systémové hypotenzi [28].
Efekt beraprostu je podpořen jednoznačnými daty především u nemocných ve stupni II a III funkční klasifikace podle NYHA. Nemáme data o účinnosti perorální léčby v pokročilých stadiích onemocnění. Beraprost tak zůstane zřejmě rezervován především pro pacienty v časnějších stadiích onemocnění. Byl schválen pro léčbu PAH v Japonsku.
Iloprost
Iloprost je stabilní analog prostacyklinu, který lze podávat intravenózně, perorálně a inhalačně. Kontinuální intravenózní infuze je u pacientů s PAH pravděpodobně stejně účinná jako léčba epoprostenolem, schází však větší zkušenosti. Perorální podání je limitováno rychlou degradací přípravku prostřednictvím b-oxidace ve stěně střeva a v játrech.
První zkušenosti s inhalační léčbou pocházejí z roku 1996. Inhalace iloprostu vede k výraznější akutní vazodilataci v plicním řečišti než inhalace NO. Účinek přetrvává 60–120 minut. Pro navození trvalého účinku na plicní cirkulaci je pak zapotřebí 6–12 inhalací denně. Inhalační léčba předpokládá takový charakter aerosolu, aby maximum léku dosáhlo úrovně alveolu. Optimální průměr kapének je proto 3–5 mm. Systémové účinky iloprostu jsou udávány jako zanedbatelné.
Evropská multicentrická, randomizovaná a placebem kontrolovaná studie AIR (Aerosolized Iloprost Randomized Study) zahrnula celkem 203 pacientů s idiopatickou PAH, PAH při systémových onemocněních a s chronickou tromboembolickou plicní hypertenzí ve funkčním stadiu NYHA III a IV (tab. 9). Ve skupině léčených pacientů, kteří inhalovali 6–9x denně 2,5–5 mg iloprostu, došlo po 12 týdnech k signifikantnímu zlepšení tolerance zátěže a ke zlepšení hemodynamických parametrů. Tachyfylaxe nebyla pozorována [29]. V otevřené pokračující fázi této studie bylo sledováno celkem 63 nemocných po dobu 2 let. U pacientů s idiopatickou PAH a chronickou tromboembolickou plicní hypertenzí byla prokázána stabilizace hemodynamiky a funkční zdatnosti. Přežití ve skupině idiopatické PAH bylo 91 % proti očekávaným 63 %.
Inhalace iloprostu představuje zajímavou alternativu aplikace prostacyklinu, jejíž předností je zejména minimalizace systémových účinků přípravku. Zájem se nyní soustředí na kombinaci léčby iloprostem a inhibitory fosfodiesterázy [30].
Iloprost je registrován v Evropské unii pro inhalační léčbu idiopatické a familiární PAH ve funkčním stadiu NYHA III. Intravenózní aplikace iloprostu je schválena pouze na Novém Zélandu.
Antagonisté endotelinových receptorů
Endotelin-1 je nejpotentnější endogenní vazokonstriktor produkovaný cévním endotelem, který má navíc vlastnosti mitogenní a proliferační. Endotelin-1 se váže na 2 typy receptorů, ETA a ETB. Receptor ETA je exprimován v buňkách hladkého svalstva, receptor ETB navíc ještě v buňkách endoteliálních. Aktivace receptorů ETA a ETB v buňkách hladkého svalu vede k vazokonstrikci a proliferaci. Aktivace receptorů ETB na endoteliálních buňkách aktivuje uvolnění prostacyklinu a NO.
U nemocných s PAH je pozorována vyšší sérová hladina endotelinu-1 a korelace mezi hladinou endotelinu-1 a hemodynamickými parametry [31].
V léčbě PAH se využívá duální blokády obou receptorů nebo selektivní blokády receptoru ETA. Podstatnou výhodou antagonistů endotelinových receptorů je možnost podání per os, nevýhodou pak na dávce závislá hepatotoxicita.
Bosentan
Bosentan je duální antagonista receptorů pro endotelin-1 s výraznější afinitou k receptoru ETA. Efekt léčby bosentanem u PAH je prokázán ve dvou klinických studiích, které srovnávaly bosentan a konvenční léčbu proti samotné konvenční terapii (tab. 10).
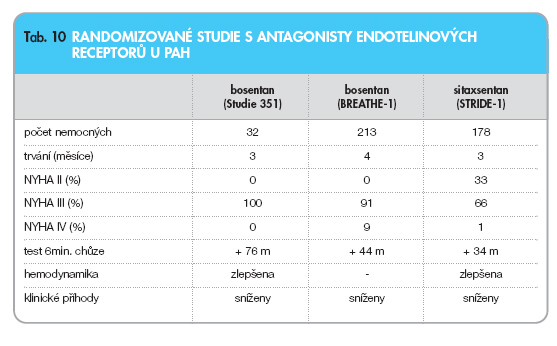
První multicentrická, dvojitě slepá a placebem kontrolovaná studie (Studie 351) randomizovala 33 nemocných s idiopatickou PAH nebo s PAH při sklerodermii ve stadiu NYHA III. V léčené skupině dostávali pacienti bosentan 2x denně v dávce 62,5 mg po dobu 4 týdnů a pak 2x denně v dávce 125 mg. U těchto nemocných došlo k signifikantnímu zlepšení tolerance zátěže a ke zlepšení hemodynamických parametrů: zvýšil se srdeční index, snížil se střední tlak v plicnici a klesla plicní cévní rezistence [32].Studie BREATHE-1 (Bosentan Randomized Trial of Endothelin Antagonist Therapy for Pulmonary Hypertension) byla dvojitě slepá, multicentrická, placebem kontrolovaná a zahrnula celkem 213 nemocných s idiopatickou PAH a PAH při sklerodermii. Zkoušely se dávky 2x denně 125 mg a 2x denně 250 mg proti placebu. U léčených pacientů došlo ke zlepšení tolerance zátěže a ke zlepšení hemodynamických parametrů. V podskupině PAH při sklerodermii byl zaznamenán především trend ke stabilizaci onemocnění. Významné zvýšení účinku v závislosti na dávce nebylo patrno, při dávkování 2x denně 250 mg byla častější indukovaná hepatopatie [33].
Echokardiografická studie v rámci BREATHE-1 sledovala změny některých echokardiografických parametrů při léčbě bosentanem. Ve skupině léčených pacientů bylo patrno zlepšení systolické funkce pravé komory a zlepšení diastolické funkce levé komory.
Studie BREATHE-2 sledovala účinek léčby bosentanem v kombinaci s epoprostenolem u 33 nemocných s plicní arteriální hypertenzí. Ukázala, že kombinovaná léčba může být účinnější než léčba samotným epoprostenolem [34].
U dětí s plicní arteriální hypertenzí ve věku 4–17 let byl bosentan zkoušen jako monoterapie, nebo v kombinaci s epoprostenolem (studie BREATHE-3). V obou případech došlo ke zlepšení hemodynamických parametrů po 12 týdnech léčby [35].
Byl rovněž prokázán účinek léčby bosentanem u PAH při infekci HIV (studie BREATHE-4).
Léčba bosentanem rovněž zásadním způsobem ovlivňuje prognózu nemocných s PAH. Při sledování 169 nemocných léčených bosentanem bylo jednoleté přežívání 96 % proti očekávaným 69 % a dvouleté přežívání 89 % proti očekávaným 57 %. jako negativní prognostický faktor byla shledána vzdálenost dosažená při testu šestiminutové chůze pod 358 m a stadium NYHA IV před léčbou [36].
K hlavním nežádoucím účinkům bosentanu patří reverzibilní a na dávce závislá hepatopatie. Vyskytuje se asi u 10–11 % léčených nemocných. Kontrola jaterních testů je nutná po 2 týdnech od zvýšení dávky a dále v měsíčních intervalech během léčby. K normalizaci testů vede redukce dávky nebo přerušení léčby. Bosentan dále vzácně navozuje anémii, interaguje s podáváním cyklosporinu A a je teratogenní. Vhodná antikoncepce u fertilních žen je při léčbě obligatorní.
Bosentan je registrován pro léčbu PAH ve stadiu NYHA III s podmínkou pravidelného monitorování jaterních testů v měsíčních intervalech.
Sitaxsentan
Sitaxsentan je prakticky selektivní antagonista receptoru ETA pro endotelin. Byla publikována randomizovaná, placebem kontrolovaná studie STRIDE-1 (Sitaxsentan To Relieve Impaired Exercise) prokazující příznivý vliv sitaxsentanu na zlepšení funkční zdatnosti, hemodynamických parametrů a redukci klinických příhod u nemocných s PAH. Problémem je indukce hepatopatie v podobném procentu případů jako u bosentanu, zejména při vyšších dávkách. Při léčbě sitaxsentanem dochází k inhibici cytochromu P-450 s nutností redukce dávky konkomitantně podávaného warfarinu [37]. V současné době probíhá další kontrolovaná studie se sitaxsentanem sledující vedle účinku především bezpečnost přípravku.
Ambrisentan
Ambrisentan je rovněž selektivní perorální antagonista endotelinových receptorů. Pilotní studie se 64 nemocnými s PAH ukázala zlepšení funkčních a hemodynamických parametrů. Lék je dále zkoušen ve dvou randomizovaných studiích.
NO a inhibitory fosfodiesterázy
NO je potentní vazodilatátor, jehož hlavním zdrojem v plicích je cévní endotel a epitel dýchacích cest. Lokální produkce NO promptně a citlivě reguluje perfuzi v závislosti na alveolární ventilaci. NO je syntetizován prostřednictvím NO syntázy z aminokyseliny L-argininu. NO zvyšuje syntézu intracelulárního cyklického guanosinmonofosfátu (cGMP), který je degradován v plicích zejména účinkem fosfodiesterázy 5 (PDE-5). Pro PAH je charakterická nedostatečná produkce endogenního NO [38].
NO
Inhalace NO vede k významné akutní vazodilataci v plicním řečišti. Dlouhodobá inhalace NO zlepšuje toleranci zátěže [39]. Technika kontinuální inhalace je však obtížná, nepraktická a vzhledem k charakteru plynu potenciálně nebezpečná. Zprávy o inhalačním podání NO v chronické léčbě PAH se v současné době omezují na kazuistická sdělení. Scházejí systematické práce, které by studovaly, zda dlouhodobá domácí inhalace NO je bezpečná a účinná. Inhalace NO se však doporučuje vedle podání adenosinu a prostacyklinu při testování akutní plicní vazoreaktivity.
L-arginin
Zprávy o intravenózním podání L-argininu jako substrátu syntézy NO jsou při popisování účinku na plicní cirkulaci u PAH velmi rozporuplné. Množí se však sdělení o krátkodobém účinku perorálně podaného L-argininu v malých nekontrolovaných studiích s PAH [40]. Výsledky randomizovaných studií scházejí. Rovněž dlouhodobý účinek perorální léčby L-argininem zůstává otázkou.
Sildenafil
Sildenafil je perorální selektivní inhibitor fosfodiesterázy 5. Je známo, že sildenafil potencuje vazodilatační účinek prostacyklinu. Na sklonku roku 2004 byly zveřejněny výsledky randomizované, placebem kontrolované studie SUPER-1 (Sildenafil Use in Pulmonary Arterial Hypertension) u nemocných s PAH ve stadiu NYHA II a III. Studie zahrnula 278 pacientů, kteří užívali 12 týdnů placebo, nebo sildenafil 3x denně v dávce 20, 40 nebo 80 mg. V léčené skupině došlo ke zlepšení vzdálenosti při testu šestiminutové chůze průměrně o 45 m bez zásadní závislosti na dávce. Rovněž se zlepšily hemodynamické parametry. Předmětem dalšího testování je dlouhodobý účinek léčby sildenafilem a efekt kombinační léčby sildenafilem a epoprostenolem.
V současné době sildenafil představuje zejména alternativu pro nemocné, u nichž selhala nebo není možná jiná schválená léčba. Po registraci přípravku v indikaci PAH lze očekávat, že se sildenafil stane pro část nemocných lékem volby [41].
Podobně jako sildenafil budou pravděpodobně v léčbě PAH účinné další inhibitory fosfodiesterázy 5 (tardalafil, vardenafil), nejsou však v této indikaci šířeji klinicky zkoušeny.
Další perspektivy farmakoterapie PAH
Vazoaktivní intestinální peptid (VIP) inhibuje agregaci trombocytů, proliferaci buněk hladkého svalstva a má významné vazodilatační účinky. Na malé skupině nemocných s PAH je dokumentováno zlepšení funkčních a hemodynamických parametrů po podání VIP [42].
V léčbě PAH se rovněž zkouší selektivní inhibitory zpětného vychytávání serotoninu (fluoxetin).
Kombinační léčba u PAH je v současné době pravděpodobně nejintenzivněji studovanou terapeutickou možností. Umožňuje postihnout více patogenetických mechanismů, které se podílejí na rozvoji onemocnění [43]. Existují 2 strategie: současné zahájení léčby dvěma nebo více přípravky, nebo přidání dalšího léku (dalších léků) k probíhající terapii. Neexistují důkazy pro to, která z těchto koncepcí je vhodnější. Máme povzbuzující zprávy o účinku kombinace bosentanu nebo sildenafilu s analogy prostacyklinu. Naopak kombinace sildenafilu a vazodilatační léčby neměla očekávaný efekt. Řada otázek zůstává zatím zcela bez odpovědi, např. problematika farmakokinetických a farmakodynamických interakcí.
Současná strategie léčby PAH
Léčba PAH se soustřeďuje na nemocné ve funkčním stadiu NYHA III a IV. Pro jasné doporučení léčebné strategie ve stadiu NYHA I a II nejsou dosud dostatečné důkazy [44, 45]. Celosvětovým standardem je soustředění diagnostiky a léčby nemocných s PAH do specializovaných center.
Nemocní s pozitivním testem akutní plicní vazodilatace mají být léčeni vysokými dávkami blokátorů kalciových kanálů. Účinek léčby je nutno ověřit průkazem funkčního a hemodynamického zlepšení po 3–6 měsících léčby. Pokud není přesvědčivý, postupuje se jako u nemocných bez vazodilatační odpovědi podle aktuálního funkčního stadia.
U pacientů bez vazodilatační odpovědi ve stadiu NYHA III, se vzdáleností dosaženou při testu šestiminutové chůze více než 330 m, je indikována léčba bosentanem nebo prostanoidy (subkutánní treprostinil, inhalační iloprost, intravenózní epoprostenol). Výběr přípravku je přísně individuální u každého pacienta. Pokud tříměsíční farmakoterapie nevede ke klinickému zlepšení, mají být pacienti zařazeni na čekací listinu k transplantaci plic.
U pacientů bez vazodilatační odpovědi ve stadiu NYHA IV, se vzdáleností dosaženou při testu šestiminutové chůze méně než 330 m, je metodou volby kontinuální infuzní léčba epoprostenolem. Pro stadium NYHA IV je rovněž schválena léčba treprostinilem. Současně se zahájením farmakoterapie mají být pacienti zařazeni na čekací listinu k transplantaci plic. Lze je vyřadit v případě zlepšení a stabilizace stavu ve stadiu NYHA II a opět zařadit při zhoršení o 1 stupeň NYHA. Ve stadiu NYHA IV může být u některých nemocných při splnění přísných indikačních kritérií k překlenutí doby do transplantace vhodná perkutánní balonková atriální septostomie.
Závěr
PAH zůstává nadále nevyléčitelným onemocněním. Současná farmakoterapie dokáže zmírnit symptomy onemocnění, ovlivnit hemodynamické parametry a snad také zlepšit kvalitu života a prognózu nemocných. Díky významné ekonomické náročnosti je však dostupnost léčby limitovaná. Navíc zejména léčba prostanoidy je značně komplikovaná řadou nežádoucích účinků plynoucích ze způsobu podání. Velkou nadějí je proto existence perorální terapie. Ta je však ve svém účinku dávkou limitovaná a při progresi onemocnění se může stát neúčinnou. Jako velmi perspektivní se proto jeví kombinační léčba. Další studium farmak u PAH bude v řadě ohledů komplikované. Významným faktorem je limitovaný počet nemocných pro vytváření rozsáhlých randomizovaných studií. Placebem kontrolované studie se stanou z etického hlediska postupně zřejmě neschůdnými pro známou rychlou progresi neléčeného symptomatického onemocnění.
Kvůli vzácnosti plicní arteriální hypertenze, komplikovanosti jejího diagnostického určení a náročnosti léčby je nezbytností a celosvětovým trendem soustředit péči o tyto pacienty do specializovaných center.
Seznam použité literatury
- [1] Riedel M. Klasifikace a nomenklatura plicní hypertenze. Kapitoly z kardiologie 2002; 4: 46–49.
- [2] Simonneau G, Galie N, Rubin LJ, et al. Clinical classification of pulmonary hypertension. J Am Coll Cardiol 2004; 43: 5S–12S.
- [3] Barst RJ, McGoon M, Torbicki A, et al. Diagnosis and differential assessment of pulmonary arterial hypertension. J Am Coll Cardiol 2004; 43: 40S–47S.
- [4] Archer S, Rich S. Primary pulmonary hypertension. Circulation 2000; 102: 2781–2791.
- [5] Kuo PC, Plotkin JS, Gaine S, et al. Portopulmonary hypertension and the liver transplant candidate. Transplantation 1999; 67 (8): 1087–1093.
- [6] Mehta NJ, Ijaz KA, Rajal N, et al. HIV-related pulmonary hypertension analytic review of 131 cases. Chest 2000; 118: 1133–1141.
- [7] Abenhaim L, Moride Y, Brenot F, et al. Appetite-suppressant drugs and the risk of primary pulmonary hypertension. N Engl J Med 1996; 335 (9): 609–616.
- [8] Farber HW, Loscalzo J. Pulmonary arterial hypertension – mechanism of disease. N Engl J Med 2004; 351: 1655–1665.
- [9] Nichols WC, Koller DL, Slovis B, et al. Localization of the gene for familial primary pulmonary hypertension to chromosome 2q31-32. Nat Genet 1997; 15 (3): 277–280.
- [10] Fishman AP. Etiology and pathogenesis of primary pulmonary hypertension. Chest 1998; 114: 242S–247S.
- [11] D´Alonzo GE, Barst RJ, Ayers SM, et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med 1991; 115: 343–349.
- [12] McLaughlin VV, Presberg KW, Doyle RL, et al. Prognosis of pulmonary arterial hypertension. ACCP evidence-based clinical practice guidelines. Chest 2004; 126: 78S–92S.
- [13] Rubin LJ. Primary pulmonary hypertension. Chest 1993; 104 (1): 236–250.
- [14] Badesch DB, Abman SH, Ahearn GS. Medical therapy for pulmonary arterial hypertension. ACCP evidence-based clinical practice guidelines. Chest 2004; 126: 35S–62S.
- [15] Galie N, Seeger W, Naeije R, et al. Comparative analysis of clinical trials and evidence-based treatment algorithm in pulmonary arterial hypertension. Am Coll Cardiol 2004; 43: 81S–88S.
- [16] Sitbon O, Humbert M, Ioos V, et al. Who benefits from long-term calcium-channel blocker therapy in primary pulmonary hypertension? Am J Respir Crit Care Med 2003; 167: A 440.
- [17] Rich S, Kaufmann E, Levy PS. The effect of high doses of calcium channel blockers on survival in primary pulmonary hypertension. N Engl J Med 1992; 327: 76–81.
- [18] Fuster V, Steele PM, Edwards WD, et al. Primary pulmonary hypertension and the importance of thrombosis. Circulation 1984; 70: 580–587.
- [19] Klings ES, Farber H. Current management of primary pulmonary hypertension. Drugs 2001; 61: 1945–1956.
- [20] Galie N, Torbicki A, Barst R, et al. Guidelines on diagnosis and treatment of pulmonary arterial hypertension. The task force on diagnosis and treatment of pulmonary arterial hypertension of the European society of cardiology. Eur Heart J 2004; 25: 2243–2278.
- [21] Tuder RM, Cool CD, Geraci MW, et al. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension Am J Respir Crit Care Med 1999; 159: 1925–1932.
- [22] Barst RJ, Rubin LJ, Long WA, et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med 1996; 334: 296–301.
- [23] Rubin LJ, Mendoza J, Hood M, et al. Treatment of primary pulmonary hypertension with continuous intravenous prostacyclin (epoprostenol). Results of a randomized trial. Ann Intern Med 1990;112: 485–491.
- [24] Badesch DB, Tapson VF, McGoon MD, et al. Continuous intravenous epoprostenol for pulmonary hypertension due to the scleroderma spectrum of disease. A randomized, controlled trial. Ann Intern Med 2000; 132: 425–434.
- [25] Simonneau G, Barst RJ, Galie N, et al. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: a double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med 2002; 165: 800–804.
- [26] McLaughlin VV, Gaine SP, Barst RJ et al. Efficacy and safety of treprostinil, an epoprostenol analog for primary pulmonary hypertension. J Cardiovasc Pharmacol 2003; 41: 293–239.
- [27] Galie N, Humbert M, Vachiery JL et al. Effects of beraprost sodium, an oral prostacyclin analogue, in patients with pulmonary arterial hypertension: a randomized, double-blind, placebo-controlled trial. J Am Coll Cardiol 2002; 39: 1496–1502.
- [28] Barst RJ, McGoon, McLaughlin VV, et al. Beraprost therapy for pulmonary arterial hypertension. J Am Coll Cardiol 2003; 41: 2119–2125.
- [29] Olschewski H, Simonneau G, Galie N, et al. Inhaled iloprost for severe pulmonary hypertension. N Engl J Med 2002; 347: 322–329.
- [30] Ghofrani HA, Rose F, Schermuly RT, et al. Oral Sildenafil as long-term adjunct therapy to inhaled iloprost in severe pulmonary arterial hypertension. J Am Coll Cardiol 2003; 42:158–164.
- [31] Stewart DJ, Levy RD, Cernacek P, et al. Increased plasma endothelin-1 in pulmonary hypertension: marker or mediator of disease? Ann Intern Med 1991; 114: 464–469.
- [32] Channick R, Badesch DB, Tapson VF, et al. Effects of the dual endothelin receptor antagonist bosentan in patients with pulmonary hypertension: a placebo-controlled study. J Heart Lung Transplant 2001; 20: 262–263.
- [33] Rubin LJ, Badesch DB, Barst RJ, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med 2002; 346: 896–903.
- [34] Kim NS, Channick R, Rubin LJ. Successful with-drawal of chronic epoprostenol therapy for pulmonary arterial hypertension. Chest 2003; 24: 1612–5.
- [35] Barst RJ, Ivy D, Dingemanse, et al. Pharmacokinetics, safety, and efficacy of bosentan in pediatrics patiens with pulmonary arterial hypertension. Clin Pharmacol Ther 2003; 73: 372–82.
- [36] McLaughlin VV, Sitbon O, Badesch DB, et al. Survival with first-line bosentan in patients with primary pulmonary hypertension. Eur Respir J 2005; 25: 244–249.
- [37] Barst RJ, Langleben D, Frost A, et al., and the STRIDE-1 Study Group. Sitaxsentan therapy for pulmonary arterial hypertension. Am J Respir Crit Care Med 2004; 169: 441–447.
- [38] Giaid A, Saleh D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med 1995; 333: 214–221.
- [39] Hasuda T, Satoh T, Shimuchi A, et al. Improvement in exercise capacity with nitric oxide inhalation in patients with precapillary pulmonary hypertension. Circulation 2000; 101: 2066–2070.
- [40] Nagaya N, Uematsu M, Oya H, et al. Short-term oral administration of L-arginine improves hemodynamics and exercise capacity in patients with precapillary pulmonary hypertension. Am J Respir Crit Care Med 2001; 163: 887–891.
- [41] Ghofrani HA, Pepke-Zaba J, Barbera JA, et al. Nitric oxide pathway and phosphodiesterase inhibitors in pulmonary arterial hypertension. J Am Coll Cardiol 2004; 43: 68S–72S.
- [42] Petkov V, Mosgoeller W, Ziesche R, et al. Vasoactive intestina peptide as a new drug for treatment of primary pulmonary hypertension. J Clin Invest 2003; 111: 1339–1346.
- [43] Hoeper MM, Dinh-Xuan AT. Combination therapy for pulmonary arterial hypertension: still more questions than answers. Eur Respir J 2004; 24: 339–340.
- [44] Galie N, Seeger W, Naeije R, et al. Comparative analysis of clinical trials and evidence-based treatment algorithm in pulmonary arterial hypertension. J Am Coll Cardiol 2004; 43: 81S–88S.
- [45] Jansa P, Aschermann M, Riedel M, et al. Doporučení pro diagnostiku a léčbu plicní arteriální hypertenze v ČR. Cor Vasa 2004; 46 (3): K35–K44.