Osa renin-angiotenzin-aldosteron – půl století od objasnění funkce a stále nová překvapení
Během vývoje musely živočišné druhy čelit měnícím se životním podmínkám. Na tyto změny bylo nutno se adaptovat. Tak například v době, kdy primitivní obojživelníci opouštěli mateřské prostředí a vydávali se z moře na souš, vznikly v souvislosti se změnou životního prostředí nové regulační systémy, které umožnily přežití na souši – zajistily udržení vnitřního prostředí a krevního tlaku v nových podmínkách. Objevuje se osa renin-angiotenzin-aldosteron (RAA) a paralelní systémy (např. kininový či apelinový), které kontrolují činnost tohoto vazokonstrikčního, natrium-retenčního a zánět aktivujícího systému. Bohužel, v řadě patologických situací, jakými jsou např. hypertenze, srdeční selhání či diabetes, je systém v nerovnováze, posunut na stranu vazokonstrikce, volumové expanze a zánětlivé reakce. Nadměrně stimulována je právě osa RAA a tato nežádoucí hyperaktivita výrazně akceleruje progresi kardiovaskulárních chorob. Z těchto důvodů je nutno systém tlumit. Vzhledem k tomu, že zmíněné choroby jsou přítomny nejméně u 40 % populace naší kulturní oblasti, stojí léky tlumící aktivitu osy RAA, tj. inhibitory ACE, sartany, inhibitory reninu či inhibitory mineralokortikoidních receptorů, v popředí zájmu. Další perspektivní léčebné postupy cílené na osu RAA či na systémy sdružené jsou ve fázi klinického hodnocení. Zmiňme jen analoga angiotenzinu 1-7, aktivátory apelinového systému, inhibitory Rho-kinázy či inhibitory aldosteron-syntázy. Nyní, po 50 letech od objasnění základního schématu mechanismu působení reninu a angiotenzinu II, je podán přehled nových poznatků, jak optimálně obnovit porušenou rovnováhu; v neposlední řadě chce tento článek informovat o tom, co se chystá ve vývojových laboratořích farmaceutických koncernů.
Proč právě přehled o systému renin-angiotenzin-aldosteron? Důvody jsou nejméně tři. První je historický – uplynulo přesně půl století od doby, kdy Skeggs se spolupracovníky objasnil základní schéma fungování osy a kdy Gross vysvětlil vztah k aldosteronu. Stejně tak dlouho trvalo, než došlo od prvé Tiegerstedtovy demonstrace účinku reninu na krevní tlak k tomuto zásadnímu objevu [1–2]. Nabízí se otázka, zda ona půlstoletí, která můžeme na pomyslné časové ose vyznačit oběma směry, byla stejně plodná. Druhým důvodem je recentní zavedení prvého přímého inhibitoru reninu – aliskirenu – do klinické praxe. Paradoxem je, že tuto cestu nastínil právě před padesáti lety Skeggs jako nejperspektivnější. Výzkum však směřoval nejprve k vývoji inhibitorů ACE a blokátorů receptorů AT1 čili sartanů. Jako třetí důvod bychom mohli uvést zveřejnění výsledků studie ONTARGET, která porovnávala – dosud nejrozsáhleji – efekt podávání sartanů, inhibitorů ACE či jejich kombinace na blokádu systému renin-angiotenzin-aldosteron (RAA) [3].
Ač všechny zmíněné důvody jsou jistě nepřehlédnutelné, hlavním podnětem pro sepsání tohoto přehledu bylo překvapivé množství zajímavých informací, o něž byly naše znalosti v posledních letech obohaceny. Pokrok v základním výzkumu bývá zpravidla vzápětí následován i zásadními inovacemi léčebných postupů. Podívejme se proto, jak bylo využito těchto znalostí doposud a jaké jsou další perspektivy.
Proč takový zájem o systém renin-angiotenzin-aldosteron?
Uvědomme si, že největších pokroků v léčbě kardiovaskulárních chorob bylo dosaženo zdánlivě paradoxním způsobem – blokádou pochodů a regulačních mechanismů, které jsou zodpovědné za udržení homeostázy a za správnou funkci organismu. Máme na mysli inhibici pochodů chronicky hyperaktivovaných, které mají při svém dlouhodobém působení maladaptační charakter. Filozofie vývoje vychází totiž z pravidla, že v rámci fylogeneze slouží veškeré regulační mechanismy tak, aby byly výhodné pro akutně ohroženého jedince, tj. ohroženého bojem (např. akutním krvácením) či dočasnými nepříznivými životními podmínkami (např. spojenými s dehydratací či s hladověním). Naopak chronicky nemocného tento fylogenetický tlak jasně eliminuje jako jedince neperspektivního. Je to až naše civilizace, která se tomuto zákonu postavila a stará se i o své nejslabší články, tj. lidi chronicky nemocné.
Regulačních mechanismů bylo během fylogeneze vyvinuto mnoho. Některé sloužily pouze u nižších živočichů a s vývojem přestaly být uplatňovány, jiné se funkčně dochovaly u druhu Homo sapiens dodnes a uplatňují se i ve zcela změněných podmínkách „homo sedativus", člověka sedavého, či „homo obesus", člověka obézního. Právě tyto nefyziologické podmínky vedou k maladaptačnímu působení dvou základních regulátorů: systému sympatoadrenálního a systému RAA. Prvý je aktivován při akutním stresu, např. při boji či útěku, druhý při déletrvajících nepříznivých podmínkách. Stimulace sympatiku je součástí fyziologické obranné reakce, v popředí je vazokonstrikce, zvýšení kontraktility, zrychlení srdeční frekvence, aktivace hemostázy, tedy reakce směřující k udržení oběhu. Navíc je stimulována plejáda metabolických změn zvyšujících nabídku rychle využitelných energetických zdrojů. To vše vede, při krátkodobém trvání, ke zlepšení vyhlídek na přežití. Naopak trvá-li zvýšený tonus sympatiku za patologických stavů dlouhodobě – např. u srdečního selhání, diabetu, hypertenze, fibrilace síní, myokardiální ischémie či při iktu – je tato hyperaktivace nežádoucí. Chronická vazokonstrikce vede k hypertenzi, zvýšená sarkoplazmatická hladina kalcia k elektrické nestabilitě myokardu s rizikem vzniku arytmií. Paralelně dochází k aktivaci trombocytů a ke zvýšení hladiny fibrinogenu, což nastartuje primární i sekundární hemostázu s následnými trombotickými a tromboembolickými komplikacemi, a stresová aktivace metabolismu glycidů a lipidů navodí komplex nežádoucích změn ústících v akceleraci aterogeneze a trombogeneze.
Zcela obdobně je krátkodobá aktivace systému renin-angiotenzin-aldosteron výhodná při akutním ohrožení – vazokonstrikce a retence tekutin a soli udrží dostatečný krevní tlak i zásobení životně důležitých orgánů, aktivace zánětlivých pochodů umožní včasnou reparaci poškozených tkání a aktivace hemostázy sníží riziko krvácení. I zde platí, že za patologických situací, jakými jsou hypertenze, srdeční selhání, diabetes, fibrilace síní nebo myokardiální ischémie, je systém dlouhodobě stimulován a je nutno jej tlumit. Negativní dopad chronické hyperaktivace osy RAA se projeví v ještě širší paletě maligních změn. Na úrovni řízení krevního tlaku je stimulována retence natria a vody v ledvinách navozená jak angiotenzinem II (AII), tak mineralokortikoidním efektem aldosteronu. Navíc působením AII na osmoreceptory v mozku je navozen pocit žízně. Vazokonstrikce je v bezprostředním horizontu výsledkem přímého působení AII na hladkou svalovinu. Trvá-li však hyperstimulace AII déle, dojde k vývoji endoteliální dysfunkce, kdy selhávají vazodilatační mechanismy (zejména klesá dostupnost oxidu dusnatého), až posléze přestavba cévní stěny při proliferaci vaziva a hypertrofii svaloviny přispívá k dlouhodobému zvýšení periferního odporu a tím i k fixaci hypertenze. Výsledný vzestup krevního tlaku je pak souhrou zvýšení cirkulující tekutiny (volum-dependentní složka) i nadměrného periferního odporu při vazokonstrikci (rezistenční složka). Dopad na metabolismus vyplývá ze snížení senzitivity k inzulinu s nežádoucí hyperinzulinémií a z facilitace transportu cholesterolu do tkání cestou LDL, v neposlední řadě pak i ze zvýšení oxidačního stresu v cévní stěně. Aterogenní efekt hyperaktivace RAA má podklad nejen ve vzestupu tlaku a v metabolickém efektu, ale i v aktivaci řady mitogenně a prozánětlivě působících cytokinů (IL-6 – interleukin 6, TGF-b≤ – transforming growth factor b, PDGF – platelet-derived growth factor aj.) a vazoadhezivních molekul či selektinů. Neméně důležité je i navození protrombotického stavu, na kterém se podílí jak aktivace trombocytů, tak inhibice fibrinolýzy (zvýšením nabídky přirozeného inhibitoru aktivátoru plazminogenu – PAI-1).
Shrneme-li, jak nezdravý způsob života s nadměrným stresem, nedostatkem pohybu, obezitou, tak plejáda nejčastěji se vyskytujících onemocnění kardiovaskulárních (srdeční selhání, hypertenze, fibrilace síní, ICHS) i nekardiovaskulárních (diabetes, řada nefropatií i hepatopatií) jsou provázeny nežádoucí nadměrnou stimulací regulačních mechanismů, zejména však systému sympatoadrenálního a osy RAA. Tato hyperaktivita přispívá k rozvoji hypertenze, aterotrombózy, dysrytmií (zejména fibrilace síní), diabetu (a jeho komplikací), nefropatií (zejména nefropatie diabetické) či steatózy jater. Z těchto důvodů je potřeba oba souběžně působící systémy brzdit. Proto je náš zájem upřen na strategii jejich útlumu a proto jsou farmaka ze skupiny inhibitorů systému RAA a blokátorů sympatoadrenální aktivace nejrozšířenějšími léčivy v kardiologii a jedněmi ze skupin s nejdynamičtěji rostoucí spotřebou.
Co nového v názorech na osu RAA?
Základní působení osy RAA včetně farmakologického ovlivnění na jednotlivých etážích uvádí schéma (obr. 1). Od doby, kdy Skeggs a spolupracovníci objasnili základní osu, tj. angiotenzinogen-angiotenzin I a angiotenzin II, byl postupně objasněn vztah AII k aldosteronu, význam enzymu konvertujícího angiotenzin I (ACE) v degradaci natriureticky a vazodilatačně působícího bradykininu (obr. 1). V dalších desetiletích byl blíže prostudován význam natriuretických vazodilatačních peptidů (ANP, BNP a dalších) z myokardu srdečních síní i komor a význam jejich stimulace AII. Podobně byla vysvětlena úloha jednotlivých receptorů pro angiotenzin II, zejména podtypů AT1, AT2 a AT4.
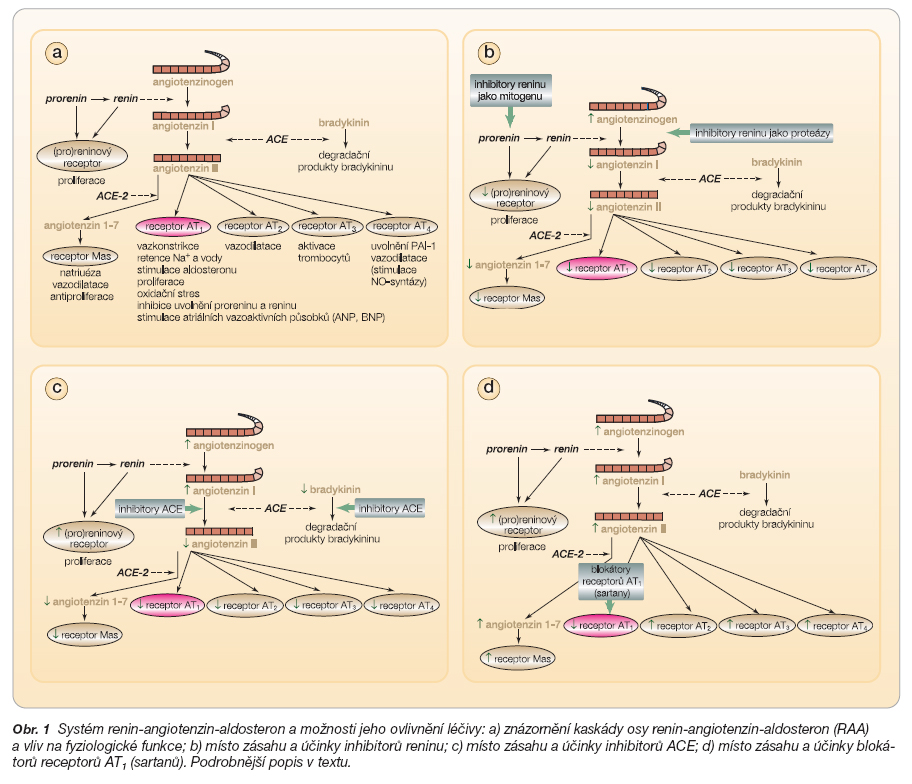
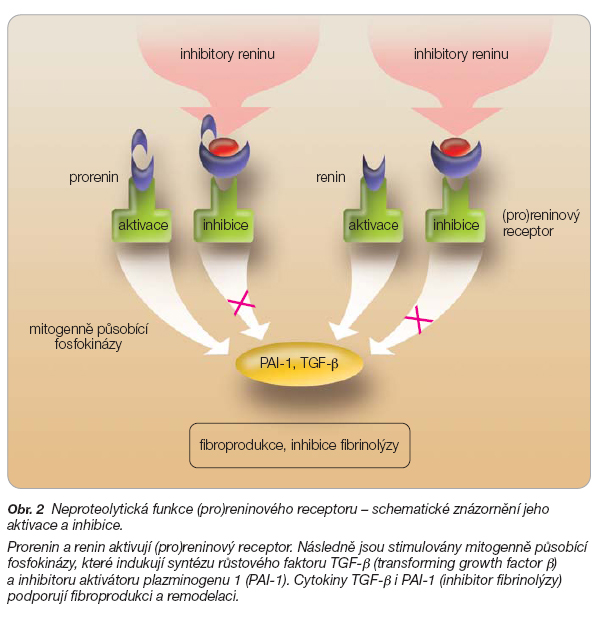
Nicméně i v průběhu posledního desetiletí došlo k objevům zásadního významu – blíže byla specifikována úloha řady „netradičních" oligopeptidů vznikajících z mateřského dekapeptidu angiotenzinu I či z oktapeptidu angiotenzinu II. Další zajímavostí jsou nové poznatky o reninu. Tento peptid nepůsobí jen „klasicky", tj. jako proteáza konvertující angiotenzinogen na AI, ale též, spolu s proreninem, jako hormon stimulující mitogenní (pro)reninový receptor (obr. 2).
Velmi zajímavou oblastí, kde došlo k vývoji našich názorů, je intrarenální působení systému a význam tkáňové aktivity AII. Zcela se též přehodnotila úloha reninu, AII i paralelních peptidů (angiotenzinu 1-7 aj.) v reparačních a zánětlivých pochodech. Konečně nutno zmínit vývoj v paralelních kontrolních systémech – kininovém (hlavním představitelem je bradykinin) a ve skupině natriuretických peptidů (zejména ANP, BNP a adrenomedulin) –, jejichž hlavní úlohou je protiváha systému RAA (navození vazodilatace, natriurézy a upravení funkce endotelu). Dalším velmi zajímavým komplexem je apelinový systém, jehož receptor (angiotensin receptor like 1) je velmi podobný receptoru AT1 či bradykininovému receptoru (vše jsou G-proteiny s obdobnou sekvencí aminokyselin). Tento systém se účastní kontroly kontraktility myokardu, napětí hladké svaloviny cévní stěny i zažívacího traktu, vylučování elektrolytů a vody či regulace glycidového a lipidového metabolismu.
Renin a prorenin jako hormon
Podíváme-li se blíže na nové poznatky o ose RAA, bude logické začít první klíčovou substancí, kterou je renin. Nejedná se pouze o monospecifickou proteázu aktivující AI, jak byl renin klasicky chápán, ale o klíčový krok limitující rychlost aktivace celého systému. Pokles krevního tlaku (cestou renálních baroreceptorů), snížení koncentrace NaCl (cestou chemoreceptorů v macula densa) nebo stimulace adrenergních receptorů b≤1 vedly k uvolnění reninu, který konvertoval angiotenzinogen na angiotenzin I. Vlastní konverze je zcela pod kontrolou reninu, v tomto kroku je jeho úloha nezastupitelná jinou proteázou. Prorenin pak byl pokládán pouze za prekurzor reninu. V posledních letech však vyvstává nová koncepce – prorenin a renin jako samostatné působky, resp. hormony. O hormonech můžeme mluvit proto, že renin, resp. prorenin má svůj orgán vzniku, má svůj receptor a má i cílovou buňku, k níž je transportován krevní cestou.
Prorenin, jehož koncentrace je asi o řád vyšší nežli koncentrace reninu, aktivuje spolu s reninem (pro)reninový receptor. Tento termín má vyjadřovat možnost vazby obou aktivátorů. Vlastní vazba reninu na receptor zvyšuje jeho enzymatickou aktivitu (tj. konverzi angiotenzinogenu na AI) asi pětinásobně. Paralelně však je vazbou (pro)reninu i reninu aktivována neproteolytická funkce receptoru (obr. 2), tj. jsou stimulovány dvě mitogenně působící fosfokinázy (p42/p44 MAPK a p38 MAPK), které indukují syntézu růstového faktoru TGF-b≤ (transforming growth factor b) a inhibitoru aktivátoru plazminogenu 1 (PAI-1). Tato aktivace je nezávislá na funkci reninu jako proteázy a není ovlivnitelná ani inhibitory ACE, ani sartany. Úloha obou cytokinů TGF-b≤ i PAI-1 v etiopatogenezi kardiovaskulárních onemocnění je klíčová, oba se účastní zejména při fibroprodukci a remodelaci.
Růstový faktor TGF-b (transforming growth factor b) je příkladem multifunkčního cytokinu akutní fáze – kontroluje produkci a degradaci extracelulární matrix, zejména tvorbu kolagenu, stimuluje hypertrofii kardiomyocytu, podporuje aktivitu lymfocytů, a naopak tlumí makrofágy, na úrovni endotelií tlumí syntézu cytokinů typu proteinů akutní fáze a vazoadhezivních molekul. V etiopatogenezi kardiovaskulárních chorob se pak uplatní na jedné straně příznivě – stabilizací aterosklerotického plátu, na druhé straně stimuluje vznik restenózy po cévní angioplastice, podílí se významně na remodelaci komor po infarktu myokardu, na fibroprodukci v srdečních předsíních s rizikem vzniku fibrilace či flutteru síní a na řadě dalších stavů spojených s vývojem fibrózy a remodelace. Aktivace osy RAA stimuluje faktor TGF-b cestou (pro)reninových receptorů i přímým působením angiotenzinu II. Blokáda aktivace TGF-b inhibitory reninu je tak dokonalejší a postihuje obě fyziologicky se uplatňující cesty aktivace [4, 5].
Druhým proteinem akutní fáze je inhibitor aktivátoru plazminogenu 1 (PAI-1). Také tento cytokin má pleiotropní efekt. Vedle klasické role přirozeného inhibitoru fibrinolýzy (tlumí efekt aktivátoru tkáňového plazminogenu – t-PA) se PAI-1 významně zapojuje v zánětlivých procesech (aktivuje proliferaci a migraci buněk účastnících se zánětlivých a reparačních pochodů). Zcela novým pohledem však je PAI-1 jako klíčový faktor metabolického syndromu, kdy se zdá, že tento faktor stojí v pozadí vývoje inzulinorezistence. Přeneseme-li zájem na dopad hyperaktivace PAI-1 na etiopatogenezi kardiovaskulárních chorob, je prokázán vliv v rozvoji aterotrombózy i v časných či pozdních stadiích aterogeneze.
Uvědomíme-li si, že podávání inhibitorů ACE i sartanů je spojeno se zvýšenou aktivitou proreninu i reninu a s hyperaktivací (pro)reninových receptorů, může zavedení nové skupiny inhibitorů reninu být velmi zajímavé. Docílíme tak nejen snížení aktivace jednotlivých receptorů pro angiotenzin II, ale i nežádoucí aktivaci (pro)reninových receptorů [6, 7]. Inhibitory reninu jako nová léková skupina tak tlumí nejen aktivitu reninu jako proteázy (obr. 1), ale i jako hormonu (obr. 1, 2). Podrobněji bude skupina probrána později.
Význam paralelních peptidů kontrolujících efekt angiotenzinu I a II – efekt nezávislý na AII
Angiotenzin II je sice hlavním aktivním peptidem osy RAA, nicméně řada souběžně vznikajících oligopeptidů má rovněž svoji fyziologickou úlohu. Za situace, jakou je blokáda receptoru AT1, kdy nabídka AI či AII výrazně stoupá, může být konverze na tyto netradiční působky podstatná. Vzhledem k tomu, že AII je velmi mohutným vazokonstrikčním, volum-expanzivním, protrombotickým působkem, aktivujícím i zánětlivé proteiny akutní fáze, není neobvyklé, že paralelně budou existovat systémy, které kontrolují jeho efekt. Jeden z těchto systémů je založen na analogických oligopeptidech obdobných angiotenzinu I či II, na které jsou tyto základní proteiny konvertovány. Jejich produkce je významná zejména v tkáních s vysokou aktivitou aminopeptidáz, jako jsou ledviny či mozek.
V CNS se uplatňuje například angiotenzin III (AIII), heptapeptid – angiotenzin 2-8, který vzniká odštěpením prvé N-terminální aminokyseliny. Je vysoce pravděpodobné, že se podílí na cirkadiálním rytmu krevního tlaku a při nadměrné koncentraci na fixaci hypertenze. V periferii navodí vazokonstrikci a proliferaci vaziva v cévní stěně. Další degradací z N-terminálního konce vzniká angiotenzin IV (angiotenzin 3-8), který stimulací NO-syntázy a zvýšením nabídky oxidu dusnatého (NO) navodí vazodilataci a potenciací efektu AII naopak podporuje vazokonstrikci. Výsledný efekt tohoto hexapeptidu je pak závislý na aktuální aktivitě AII v mozku [8, 9].
Angiotenzin I i angiotenzin II mohou být biodegradovány rovněž z C-terminálního konce. Významnou biologickou aktivitu vyvíjí heptapeptid – angiotenzin 1-7 (A1-7), který působí jako fyziologický antagonista angiotenzinu II (obr. 1). Biokonverze AI a AII na A1-7 je realizována více karboxypeptidázami (zejm. endopeptidázami či neprilysinem). Pro konverzi z AI a zejména z AII je nejvýznamnější strukturální homolog ACE, označovaný jako angiotenzin konvertující enzym 2 (ACE-2). Tento multipotentní enzym má velký biologický význam, působí jako klíčový enzym v paralelním řetězci oligopeptidů kontrolujících účinek angiotenzinu II. Na rozdíl od ACE nekonvertuje AI na AII a jeho účinek není ovlivněn inhibitory ACE. I když je zřejmé, že primárním substrátem pro ACE-2 je AII, tento enzym zaujímá významné místo i v metabolismu kininů a v apelinovém systému. I zde účinné působky (apelin-13 a aktivní fragmenty bradykininu) antagonizují vazokonstrikční efekt angiotenzinu II. V této souvislosti stojí za pozornost, že transmembránový enzym ACE-2 je jediným receptorem, jehož pomocí se do buněk respiračního systému dostávají koronaviry a způsobí tam akutní respirační postižení typu SARS [9–12].
Aktivace ACE-2 v ledvinách a v mozku slouží jako přirozený systém antagonizující účinek angiotenzinu II. Zvýšení hladiny AI či AII (substrátů pro ACE-2) podporuje tvorbu angiotenzinu 1-7. Podávání inhibitorů ACE zvyšuje výrazně nabídku AI, aplikace sartanů zvyšuje nabídku AII. Důsledky zvýšení konverze AI i AII na angiotenzin 1-7 potencují terapeutický účinek obou skupin.
Významným heptapeptidem, který se příznivě uplatňuje za řady patologických situací (srdeční selhání, hypertenze, fibrilace síní, remodelace), je výše zmíněný angiotenzin 1-7. Antagonismus k angiotenzinu II je zprostředkován stimulací specifických receptorů (Mas-R). Jejich aktivace v renálních tubulech vede k natriuréze, ve stěně cévní k omezení tvorby vaziva a k vazodilataci, v endotelu k úpravě funkce a v krvi ke stabilizaci destiček. Vazodilatace, stabilizace endotelu a trombocytů je dána univerzálním způsobem – aktivací NO-syntázy a zvýšením nabídky NO. Angiotenzin 1-7 je tak zcela analogický angiotenzinu II. Má svůj specifický konvertující enzym, má svůj specifický receptor a plní svoji biologickou funkci. Dle současných názorů je osa ACE-2–angiotenzin 1-7–receptor Mas významnou protiváhou účinku osy ACE-angiotenzin II-receptor AT1 (obr. 1) a doplňuje úlohu kininů, vazoaktivních natriuretických peptidů a apelinů [10, 12].
Účinek angiotenzinu 1-7 byl zatím sledován převážně na transgenních zvířatech s deficiencí ACE-2. Pozorovaný vyšší výskyt komorových tachyarytmií a pokles kontraktility komor vedl k vývoji analoga angiotenzinu 1-7 pod kódovým označením AVE0991. Nejsou však zprávy, že by výzkum tohoto přípravku překročil preklinickou fázi hodnocení [13].
Význam jednotlivých receptorů pro angiotenzin II (receptory AT)
Angiotenzin II i degradační produkty stimulují ve větší či menší míře receptory typu AT1, AT2, AT3 nebo AT4 patřící do velké rodiny G-receptorů. I když úloha receptorů AT1 je stěžejní, efekt ostatních podtypů je za určitých situací nezanedbatelný. Exprese těchto receptorů, zejména receptoru AT2, je sice u člověka v dospělosti malá, nicméně při jejich aktivaci zánětem (např. při aterosklerotickém postižení cévní stěny) se exprese zvyšuje a stimulace může mít fyziologický význam.
Efekt stimulace receptorů AT1 patří ke klasickým účinkům stimulace osy RAA. Účinek na kardiovaskulární aparát je dán vazokonstrikcí, vzestupem krevního tlaku, zvýšením kontraktility myokardu a proliferací buněk myokardu i cévní stěny. Renální účinek vyplývá ze stimulace tubulární reabsorpce sodíku, zpětnovazebné blokády uvolnění reninu a ze vzestupu intraglomerulárního tlaku. Volumová expanze je potencována aktivací centra žízně v hypotalamu. Stimulace sympatiku potencuje vzestup systémového krevního tlaku a pravděpodobně je hlavním důvodem vzestupu kontraktility levé komory. Efekt na nadledviny s vyplavením aldosteronu dále stimuluje retenci natria a vody. Konečně komplexní účinek na aktivaci proliferativních procesů v rámci zánětlivé reakce zvyšuje zánětlivou odpověď a oxidační stres. Blokáda těchto receptorů či snížení jejich stimulace má řadu příznivých účinků – pokles aterotrombotických komplikací, pokles krevního tlaku, zlepšení funkce levé komory, zlepšení funkce endotelu, zpomalení degenerativních pochodů v myokardu srdečních předsíní se snížením výskytu fibrilace síní, pokles retence natria a vody, snížení intraglomerulárního tlaku s poklesem proteinurie a zpomalením degenerace glomerulu (obr. 3) [11, 12].
Receptory AT2 jsou významné během organogeneze ve fetálním období. Kontrolují zejména vývoj ledvin a mozku. Postnatálně jejich exprese výrazně klesá a jsou významněji exprimovány pouze pod vlivem zánětlivých cytokinů. Tak se v dospělosti uplatní jen při lokální zánětlivé reakci – například při infiltraci plátu leukocyty a jeho destabilizaci mohou přispívat k udržení průtoku prostřednictvím vazodilatačního a antiproliferačního působení [10–12, 14, 15].
Zatímco účinek receptorů AT3 zůstává nejasný, předpokládá se jen aktivace trombocytů, receptory AT4 zprostředkují uvolnění PAI-1. Vedle AII tento receptor výrazně stimulují i degradační produkty – angiotenzin III a IV [15].
Aldosteron – „zapomenutý hormon"
Na zvýšení krevního tlaku působí v tandemu s AII i aldosteron. Dlouho vládla mineralokortikoidní koncepce aldosteronu – působku regulujícícho reabsorpci sodíku a vody v distálním tubulu a ve sběrném kanálku ledvin, který obdobným mechanismem reguluje reabsorpci sodíku a vody v tlustém střevě. Toto pojímání funkce aldosteronu jako „epiteliálního hormonu" se však v devadesátých letech změnilo v koncepci aldosteronu jako „hormonu kardiovaskulárního rizika".
I když je exprese mineralokortikoidních receptorů v hladké svalovině cév, v endoteliích i v kardiomyocytech výrazně nižší nežli v epiteliích ledvin a zažívacího traktu, aldosteron velmi významně ovlivňuje srdce i cévy. Charakteristické je navození infiltrace cévní stěny i perivaskulárních tkání monocyty/makrofágy cestou vyšší exprese vazoadhezivních molekul i COX-2 (tzv. aldosteronová vaskulitida), proliferace fibroblastů s fibrózou cévní stěny. Tento efekt je dán vlastním promitotickým efektem a zvýšením oxidačního stresu navozením exprese NAD(P)H oxidázy. V myokardu dominuje hypertrofie kardiomyocytů a fibrotizace stěny srdečních komor. Na celém procesu se podílí nejen stimulace mineralokortikoidních receptorů, ale i efekt receptorů (pro)reninových a receptorů AT1. Výsledkem je potenciace aterogeneze, náchylnost k destabilizaci plátu a k trombotickým komplikacím (při akutní infiltraci cévní stěny makrofágy) na straně jedné, či naopak ke stabilizaci plátu posílením fibrózní složky na straně druhé. V myokardu je stěžejní hypertrofie a remodelace srdečních komor se zhoršením trofiky tkáně a zhoršením mechaniky srdeční kontrakce. K dalším netradičním efektům aldosteronu patří centrální prohypertenzní efekt stimulací CNS a zvýšení exprese PAI-1 [16, 17].
Blokáda výdeje aldosteronu inhibicí ACE či blokádou receptorů AT1 je nedostatečná. Sekrece aldosteronu v kůře nadledvin je paralelně pod vlivem ACTH i chemoreceptorů (obr. 4).
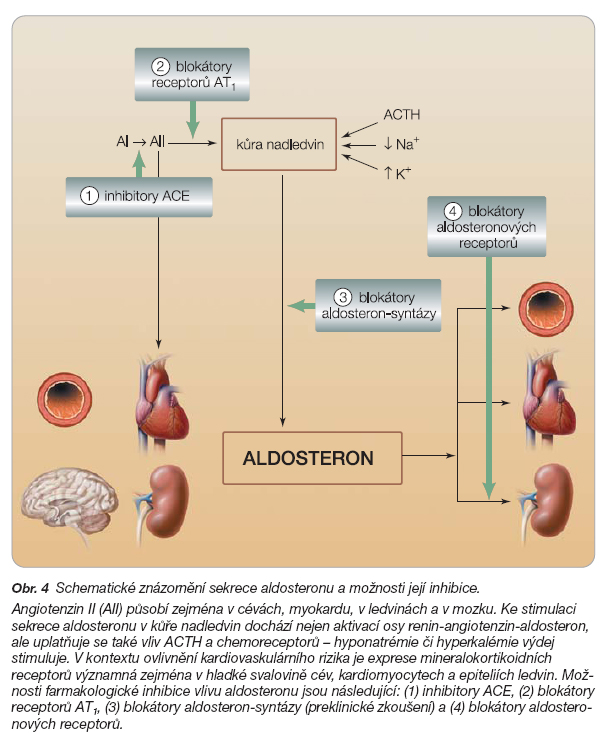
Proto optimální cestou je periferní blokáda aldosteronových (mineralokortikoidních) receptorů jejich inhibitory spironolaktonem či selektivnějším eplerenonem. Vzhledem k tomu, že periferní blokáda mineralokortikoidních receptorů vede k reciproční aktivaci celé osy RAA, je výhodné tuto skupinu kombinovat s inhibitory ACE či se sartany (obr. 4).
Ve fázi preklinického hodnocení je zajímavá skupina inhibitorů aldosteron-syntázy. Tento enzym, který stojí na konci řetězce syntézy aldosteronu, je zcela specifický pro syntézu aldosteronu a nezdá se, že by jeho blokáda ovlivňovala steroidogenezi na jiném místě. Oproti periferním blokátorům aldosteronových receptorů neaktivuje inhibice aldosteron-syntázy vlastní systém RAA [16, 17].
Kalikrein-kininový systém, další regulátor aktivity osy RAA
Kalikrein-kininový systém je obdobnou metabolickou kaskádou jako osa RAA. Její efekt na jedné straně vyvažuje retenci sodíku a vody a vazokonstrikční účinek základního systému, na straně druhé potencuje prozánětlivé působení angiotenzinu II a aldosteronu. Základní schéma bradykininové kaskády je obdobné kaskádě RAA (obr. 5) [18].
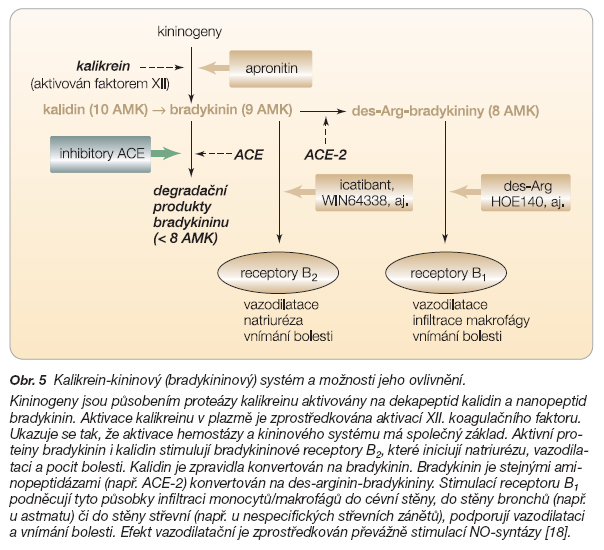
Jak vyplynulo ze studií s inhibitory bradykininových receptorů (icatibant aj.), účinek inhibitorů ACE je zprostředkován převážně zvýšením nabídky bradykininu. V kardiovaskulárním aparátu se zvýšení nabídky kininů podílí na efektu inhibitorů ACE v úpravě endoteliální dysfunkce, v antihypertenzním účinku, v antihypertrofickém a v antiproliferativním efektu. Řada studií ukazuje na spoluúčast bradykininu na příznivém efektu inhibitorů ACE v léčbě srdečního selhání. To by mohlo vysvětlit lepší efekt inhibice ACE proti inhibici receptorů AT1. Zajímavé je, že bradykinin se nepříznivě účastní na vzniku angioedému při podávání inhibitorů ACE nebo inhibitorů vazopeptidáz. Není nelogické, že blokáda receptorů B1 byla přínosná v léčbě angioedému, zejména angioedému hereditárního [19] (obr. 5).
Význam tkáňové aktivity osy RAA
V tradičním pojetí přistupujeme k systému RAA jako k hlavnímu regulátoru krevního tlaku a hospodaření s ionty a s vodou, méně tradičně jako k systému kontrolujícímu též zánětlivé a reparační pochody (včetně hemostázy). Recentně se ukazuje, že RAA ovlivňuje též diferenciaci buněk čili organogenezi a rovnováhu mezi apoptózou a proliferací tkání čili hypertrofii a její regresi. Zatímco regulace tlaku a objemu tělesných tekutin byla převážně záležitostí systémovou (až na výjimku, jakou je presorické působení terminálních aktivních proteinů v mozku a v ledvině), zásah do reparačních dějů a organogeneze je omezen převážně na lokální, tedy tkáňovou aktivitu osy. K získání komplexního pohledu na celý proces nelze proto význam tkáňového působení reninu, angiotenzinu II, angiotenzinu 1-7 a kalikreinů opominout [11, 12].
Podívejme se nejprve na význam tkáňového působení reninu. Zatímco konverze angiotenzinogenu na AI probíhá v plazmě, působení na (pro)reninový receptor je záležitostí lokální, přímo na úrovni buněčné. Prorenin i renin se z plazmy dostávají intracelulárně po glykosylaci pomocí univerzálního systému – manoso-6-fosfátového receptoru. Internalizace je nejvyšší v mozku, srdci, ledvinách a v placentě, což nasvědčuje i významu lokálního působení v jednotlivých orgánech. Vysoká aktivita (pro)reninových receptorů je v ledvině spojena s proliferací mesangia a např. u diabetické nefropatie přímo koreluje s diabetickými mikrovaskulárními komplikacemi. V ostatních orgánech není význam blíže osvětlen, pravděpodobný je vliv na organogenezi [11, 12].
Dalším významným regulátorem aktivity osy jsou konvertující enzymy: ACE a ACE-2. Jak bylo řečeno, ACE aktivuje AI na AII, zatímco ACE-2 konvertuje AII na angiotenzin 1-7. Angiotenzin konvertující enzym je aktivní v cirkulaci i v tkáních, lokální efekt angiotenzinu II se neliší významně od účinku systémového, jemuž byla věnována pozornost výše. Specifickým efektem AII, který je nutno zmínit, je indukce hypertrofie myokardu, nezávislá na výši krevního tlaku. Na receptorové úrovni jsou zapojeny receptory AT1 i AT2, které aktivují více mechanismů působících mitogenně (tyrozin-kinázu, mitogeny aktivovanou fosfokinázu, RhoA-kinázu aj.). Tato mitogenní aktivita v myokardu též stimuluje proliferaci fibroblastů, na které se podílejí nejen AII, ale též aldosteron, a výsledkem je remodelace srdečních komor. Obdobné pochody probíhají při nadměrné stimulaci systému RAA i v cévní stěně. Zde je navíc navozena stimulací receptorů AT2 angioneogeneze projevující se v myokardu zvýšenou hustotou kapilár. V mozku zmiňme pouze efekt aktivního metabolitu AII – angiotenzinu 3-8, který ovlivňuje prostřednictvím stimulace receptoru AT4 paměťové funkce (vštípivost a výbavnost). Receptory AT1 na úrovni bazálních ganglií, hypotalamu a neurohypofýzy kontrolují krevní tlak, stimulaci navodí AII, naopak útlum vyvolá například angiotenzin 1-7 [11, 12]. Na rozdíl od ACE je ACE-2 exprimován v ledvinách, v mozku, v plicích a v koronárním řečišti. Aktivita angiotenzinu 1-7 je tedy omezena na tyto orgány. Experimentální studie naznačují, že aktivita v mozku se podílí na kontrole krevního tlaku. V srdci kontroluje angiotenzin 1-7 vývoj ve fetálním období. Později tlumí vývoj hypertrofie myokardu, potencuje vazodilataci a upravuje endoteliální funkce. V tubulárním epitelu ledvin navozuje natriurézu [11, 12].
Velmi významné je též lokální působení aktivních proteinů osy RAA na úrovni reprodukce, psychických, smyslových a motorických funkcí, ledvin, zažívacího traktu i metabolismu glycidů a tuků či v prenatálním vývoji. V tomto kardiologicky orientovaném přehledu nemohou být tyto oblasti podrobněji probírány.
Možnosti inhibice osy RAA
Prvé snahy o inhibici systému RAA se datují do počátku sedmdesátých let, tj. brzy poté, co bylo zjištěno, že aktivita osy, resp. hladina reninu má negativní efekt na prognózu nemocných s hypertenzí, s ischemickými syndromy i se srdečním selháním. Prvým testovaným léčivem byl peptidový neselektivní antagonista angiotenzinových receptorů saralasin. Kvůli nutnosti parenterální aplikace a agonismu na vlastním receptoru při vyšším dávkování nepřekročilo podávání etapu klinických studií a zájem byl směřován na inhibitory ACE. Také v této skupině se nejprve testovaly oligopeptidy, nicméně již v druhé polovině 70. let se objevuje kaptopril, prvý perorálně účinný lék k blokádě osy RAA. Zavedení inhibitorů ACE do klinické praxe paradoxně vedlo k zastavení intenzivnějšího výzkumu v oblasti inhibice aldosteronových receptorů či inhibitorů reninu na řadu let. Konzultanti farmaceutické firmy Takeda, která byla v té době ve vývoji blokátorů receptorů AT1 nejdále, nepovažovali totiž tuto skupinu za perspektivní – „máme přece již účinné inhibitory ACE". Dalším paradoxem je, že trvalo plných sedm let od zavedení kaptoprilu do klinické praxe, než byla odhalena spojitost inhibitorů ACE s nejčastějším nežádoucím účinkem – kašlem. Proto se prvý blokátor receptorů AT1 – losartan – objevuje až počátkem 90. let. Mluvíme-li již o historii, dalším krokem ve vývoji byly inhibitory vazopeptidáz (VPI). Tato zajímavá skupina blokovala konverzi angiotenzinu I na angiotenzin II daleko účinněji než inhibitory ACE. Proto se předpokládalo, že inhibitory vazopeptidáz nahradí inhibitory ACE. Bohužel, inhibice i nespecifických peptidáz vedla ke zvýšení výskytu nežádoucích účinků, zejména angioedému. I když jeho výskyt ve studiích s omapatrilátem, který byl podroben zatím nejrozsáhlejšímu klinickému hodnocení ze všech inhibitorů vazopeptidáz, nebyl výrazně vyšší (0,8 % vs. 0,5 % při podávání enalaprilu), byl vývoj tohoto VPI ukončen. Nicméně vývoj v této skupině, jak se zdá, pokračuje. Ve fázi klinického hodnocení je přípravek s kódovým označením M100240 [2, 20–25].
Jaké jsou současné názory na možnosti zásahu do tohoto hlavního regulačního komplexu? Hlavním cílem v inhibici systému RAA je stále snížení stimulace hlavních receptorů angiotenzinu II, tj. podtypu AT1. Toho dosahujeme buď cestou snížení nabídky AII blokádou konvertujícího enzymu inhibitory ACE, či přímo kompetitivní blokádou tohoto receptoru blokátory receptoru AT1 čili sartany (obr. 6). Tento receptor je totiž, jak bylo již vysvětleno, u člověka nejdůležitější. Účinek obou základních skupin na regulační systém je ovšem daleko komplexnější. Předně, do hry se zapojují i ostatní působky. Jak inhibitory ACE, tak sartany zvyšují nabídku bradykininu. Ač je bradykinin často vnímán pouze jako příčina nežádoucích účinků (v případě výskytu angioedému právem, v případě výskytu kašle neprávem), skutečností je, že je významnou složkou farmakodynamického účinku obou skupin – potencuje natriurézu i vazodilataci stimulací NO-syntázy i prostaglandin-syntázy. Tímto způsobem zvyšuje nabídku dvou klíčových endoteliálních působků, oxidu dusnatého a prostaglandinu E2, a upravuje funkci endotelu. Podobně jsme dosud nezohledňovali význam ostatních biologicky aktivních peptidů, které vznikají paralelně s angiotenzinem II. Překvapivá je zejména účast angiotenzinu 1-7 jako kontrolního systému tlumícího výsledný dopad nadměrné stimulace osy RAA. Také zapojení ostatních receptorů typu AT, zejména vazodilatačních AT2 a AT4, může být při endoteliální dysfunkci a hyperaktivaci zánětlivých pochodů v cévní stěně významné [20–25].
Srovnáme-li efekt inhibitorů ACE se sartany, jsou patrné rozdíly v účinku na hlavní receptor i v dopadu na paralelní systémy. Inhibitory ACE totiž konverzi AI na AII neinhibují úplně; za patologických situací, například u srdečního selhání či při akutní stresové situaci, se v konverzi ve větší míře uplatňují i nespecifické proteázy (chymáza aj.). V důsledku toho nemusí být aktivita osy RAA utlumena inhibitory ACE dostatečně. Naopak předností inhibitorů ACE je výraznější zvýšení dostupnosti bradykininu (obr. 1c). Navíc inhibitory ACE významně stimulují bradykininové receptory typu B1 přímo a zcela nezávisle na konvertujícím enzymu [28]. Efekt na úpravu endoteliální dysfunkce, potlačení prozánětlivých cytokinů i navození vazodilatace je tak ve srovnání se sartany výraznější. Také rozdíl v efektu u srdečního selhání je vysvětlován vyšší aktivitou bradykininu, který aktivuje tzv. preconditioning, upravuje metabolismus selhávajícího myokardu a snižuje excitabilitu kardiomyocytů. Útlum ACE se projeví i nepříznivě – dochází ke zpomalení degradace tachykinů, zejména substance P, neuropeptidu Y či adrenomedulinu. Jejich vyšší aktivita stojí v pozadí hlavního nežádoucího účinku inhibitorů ACE – kašle. Jejich význam se zdá být větší nežli stimulace bronchiálních receptorů typu TRPV (Transient receptor potential vanilloid type channels) navozená bradykininem či prostaglandinem E2, které jsou považovány za příčinu kašle, zejména kašle navozeného chladným vzduchem [26–28].
Také účinek sartanů je komplexnější a nespočívá jen v blokádě receptorů AT1 (obr. 1d). Na rozdíl od inhibitorů ACE je jejich aplikace provázena vyšší nabídkou angiotenzinu II. Ten jednak stimuluje receptory AT2 a AT4, jednak je substrátem pro angiotenzin konvertující enzym typu 2 a zvýšení nabídky angiotenzinu 1-7 přispívá k farmakodynamickému efektu. Tento paralelní duální efekt přispívá k vazodilataci, k natriuréze i k protizánětlivému působení sartanů. Fakt, že efekt inhibitorů ACE na bradykinin a efekt sartanů na ostatní receptory AT i na angiotenzin 1-7 je klinicky významný, dokumentují studie s použitím blokátorů jednotlivých receptorů, blokátorů konvertujících enzymů či studie v prostředí bez ACE. Též rozdíly v účinku u srdečního selhání nelze vysvětlit jiným způsobem.
Blokáda systému RAA přinesla významné zlepšení našich léčebných přístupů. Příznivý účinek inhibitorů ACE i sartanů je dostatečně doložen v řadě důležitých indikací. V léčbě hypertenze je dokumentován nejen pokles krevního tlaku, ale i snížený výskyt kardiovaskulárních příhod a úmrtí. Účinnost inhibitorů ACE (konkrétně perindoprilu a ramiprilu) v sekundární prevenci u nemocných s projevy kardiovaskulárního či cerebrovaskulárního aterosklerotického postižení je obecně akceptována a doložena výsledky studií HOPE či EUROPA. Recentně byla výsledky studie ONTARGET doložena srovnatelná účinnost též pro sartany (resp. pro telmisartan). Také u nefropatií diabetické či jiné etiologie provázených proteinurií je dobře dokumentován pokles proteinurie i zpomalení zhoršování renálních funkcí, efekt byl srovnatelný u obou skupin. V této jediné indikaci byl navíc prokázán aditivní efekt kombinace inhibitorů ACE a sartanů. U srdečního selhání je léčba inhibitory ACE provázena zlepšením funkce levé komory i zlepšením prognózy nemocných, naproti tomu efekt sartanů je výrazně nižší. Poslední indikací, kde máme k dispozici pouze závěry metaanalýz převážně pilotních projektů a kde čekáme na výsledky velkých studií, je léčba a prevence vzniku a recidivy fibrilace či flutteru síní. I v této potenciální indikaci se zdají být obě skupiny rovnocenné.
Zaměříme-li se blíže na toleranci obou skupin, není pochyb, že hlavní předností sartanů ve srovnání s inhibitory ACE je jejich lepší tolerance, neobjevuje se kašel a výrazně nižší je výskyt angioedému. Z těchto důvodů byla u pacientů léčených sartany pozorována i větší adherence k léčbě. Výskyt nežádoucích účinků byl v kontrolovaných studiích zpravidla nižší než v placebových větvích. Společné oběma skupinám je srovnatelné riziko hyperkalémie (nutno počítat se zvýšením plazmatické hladiny draslíku v průměru o 0,5 mmol/l). Obdobný je i teratogenní efekt na vývoj urogenitálního systému v druhém trimestru gravidity [11]. Angiotenzin II se totiž podílí svým mitogenním efektem na vývoji metanefros, proto je nutno ukončit podávání inhibitorů systému RAA během prvých měsíců těhotenství.
Mluvíme-li o blokádě receptorů pro AII, nelze nezmínit velmi perspektivní cestu, jakou je inhibice Rho-kinázy. Rodina receptorů spojená s G-proteinem, kam patří i receptory AT, působí na cévy dvojím způsobem. Aktivace fosfolipázy C vede v konečném důsledku k fosforylaci lehkého řetězce myosinu a ke svalové kontrakci, stimulace Rho-kinázy snižuje hladinu oxidu dusnatého (obr. 7).
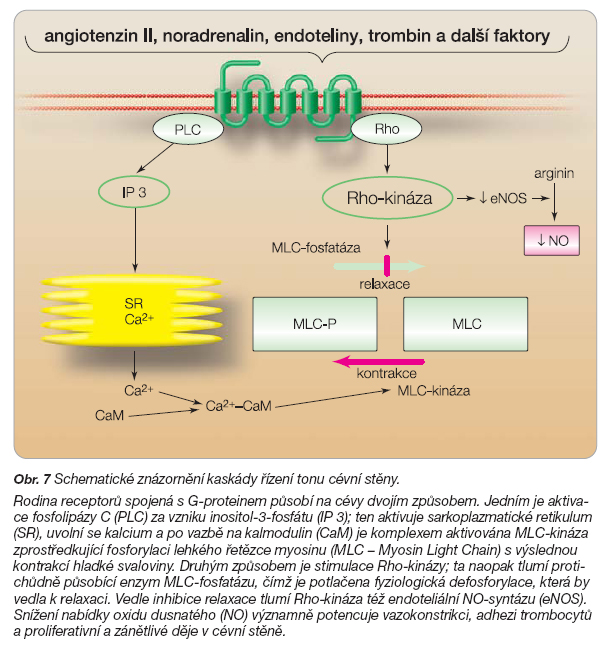
Pro tento dvojí, z hlediska etiopatogeneze kardiovaskulárních chorob nepříznivý efekt, se nabízí možnost inhibice Rho-kinázy.
První léčivo tohoto typu – fasudil – je ve fázi klinického hodnocení. Mechanismus účinku je velmi zajímavý již tím, že Rho-kináza je společnou cestou působení i jiných vazokonstrikčních hormonů a autakoidů (např. noradrenalinu či endotelinů).
Klasickou cestou inhibice osy RAA je užití blokátorů receptorů pro aldosteron. Jak již bylo uvedeno, ani sartany, ani inhibitory ACE neinhibují aldosteron dostatečně. Během posledních dvou desetiletí jsme se dopracovali ke komplexnímu chápání funkce aldosteronu. Od koncepce mineralokortikoidu podporujícího v nefronu směnu sodíku za draslík jsme se posunuli; dnes pohlížíme na aldosteron také jako na hormon působící na myokard i na cévní stěnu, kde navozuje nežádoucí proliferaci fibroblastů. Inhibice tohoto posledního článku osy blokátory aldosteronových receptorů (obr. 6) spironolaktonem či selektivněji eplerenonem má příznivý dopad na zpomalení degenerativních procesů v myokardu a ve stěně cévní.
Konečně posledním přírůstkem do rodiny inhibitorů RAA jsou přímé inhibitory reninu. V Evropské unii byl v současnosti úspěšně registrován prvý představitel skupiny – aliskiren. U zrodu této molekuly stála Alice Huxley, objevitelka, po níž získal prvou část jména. Efekt aliskirenu je po farmakologické i klinické stránce velmi zajímavý. Jak bylo uvedeno, renin není pouze proteázou, ale také působkem stimulujícím cestou (pro)reninových receptorů hypertrofii svaloviny a proliferaci vaziva cévní stěny či myokardu. Inhibitory reninu tak mají potenciál zabránit vývoji hypertrofie levé komory či navodit její regresi a snížit riziko remodelace cirkulární svaloviny arteriol a fixace zvýšené periferní rezistence. Druhým účinkem je stimulace fibrinolýzy snížením nabídky PAI-I. Na výsledném účinku se podílí útlum stimulace angiotenzinových receptorů a receptorů (pro)reninových (obr. 6). Na rozdíl od inhibitorů ACE není ovlivněna hladina bradykininu a oproti sartanům nedochází při léčbě inhibitory reninu k aktivaci angiotenzinu 1-7 a receptorů Mas [29–31].
Efekt aliskirenu byl zatím ověřen v rámci antihypertenzního působení, zajímavý je aditivní účinek k blokátorům receptoru AT1, konkrétně k valsartanu. V dalších indikacích byl ve studii ALOFT dokumentován pokles neurohumorálních působků (BNP) s negativní prognostickou vazbou u nemocných léčených inhibitory ACE či sartany. Podobně byl doložen pokles proteinurie při diabetické nefropatii, který byl rovněž aditivní ke stávající léčbě sartanem (studie AVOID). Zcela recentně byla ve studii ALLAY doložena regrese hypertrofie levé komory srovnatelná se sartany. Zatím se uskutečňují prognostické studie. Předností aliskirenu je výborný bezpečnostní profil [29–31].
Pro úplnost celého pohledu na inhibici osy RAA je potřeba uvést, že také b-blokátory inhibují tento systém (obr. 6). Jedním z jejich účinků je snížení výdeje reninu z mesangia glomerulů inhibicí stimulace receptorů b≤1. Díky tomu, že renin je uvolňován i dalšími podněty, jako jsou osmo-, chemo- či presoreceptory, je inhibice systému touto cestou nedostatečná. Nicméně i tento efekt se spolupodílí na komplexním antihypertenzním účinku b-blokátorů.
Shrneme-li, pak od roku 1957, kdy Skeggs poprvé osvětlil funkci osy RAA a navrhl inhibici reninu jako optimální krok k jeho blokádě, uplynulo pět desetiletí naplněných plodnou prací, zajímavými objevy. Základní regulační schéma renin-angiotenzin-aldosteron se rozrostlo do řady paralelních a neméně důležitých větví. V centru našeho zorného pole jsou zatím jen inhibitory ACE, sartany a blokátory aldosteronových receptorů. K dispozici jsou ale již nyní inhibitory reninu (aliskiren) nebo vazodilatační natriuretický peptid typu B (BNP – nesiritid). Perspektivní však jsou také postupy zacílené na aktivátory kininového systému, zejména bradykininu, na degradační produkty angiotenzinu, zejména angiotenzin 1-7 (testována jsou analoga tohoto fyziologického antagonisty AII). Zajímavou novinkou jsou aktivátory apelinového systému, který je analogický ose RAA, s níž má řadu styčných bodů, a který kontroluje kontraktilitu myokardu. Snad nejperspektivnější inovací budou inhibitory Rho-kinázy, klíčového enzymu, který zprostředkovává vazokonstrikci i řadu dalších funkcí nejen receptoru AT1, ale i mnoha vazokonstrikčně působících receptorů ostatních. Klinické hodnocení indikací plicní hypertenze, ischemických syndromů či hypertenze dosud nejvíce pokročilo u fasudilu; ten se zatím jeví jako nejpotentnější vazodilatátor, který byl kdy testován.
Jak je vidět, stojí před námi zajímavé roky. Jen doufejme, že hubená léta plná neúspěšných kardiologických studií již skončila. Skutečně, řada pozitivních výsledků klinických studií prezentovaných na posledním kongresu ACC na jaře 2008 (např. ONTARGET, HYVET či DUAAL) naznačuje, že se blýská na lepší časy.
Seznam použité literatury
- [1] Skeggs LT Jr, Kahn JR, Lentz K, et al. The preparation, purification, and amino acid sequence of a polypeptide renin substrate. J Exp Med 1957; 106: 439–453.
- [2] Ferrario CM. Role of angiotensin II in cardiovascular disease therapeutic implications of more than a century of research. J Renin Angiotensin Aldosterone Syst 2006; 7: 3–14.
- [3] The ONTARGET investigators: Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med 2008; 358: 1547–1559.
- [4] Verrechia F, Mauviel A. Transforming growth factor-beta and fibrosis. World J Gastroenterol 2007; 13: 3056–3062.
- [5] Bujak M, Frangogiannis NG. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res 2007; 74: 184–195.
- [6] Eddy AA, Fogo AB. Plasminogen activator inhibitor-1 in chronic kidney disease: evidence and mechanisms of action. J Am Soc Nephrol 2006; 17: 2999–3012.
- [7] Alessi MC, Juhan-Vague I. PAI-1 and the metabolic syndrome: links, causes, and consequences. Arterioscler Thromb Vasc Biol 2006; 26: 2200–2207.
- [8] Elased KM, Cunha TS, Marcondes FK, Morris M. Brain Angiotensin Converting Enzymes: Role of ACE2 in Processing Angiotensin II. Exp Physiol 2008; v tisku.
- [9] Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol 2007; 292: C82–C97.
- [10] Raizada MK, Ferreira AJ. ACE2: a new target for cardiovascular disease therapeutics. J Cardiovasc Pharmacol 2007; 50: 112–119.
- [11] Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev 2007; 59: 251–287.
- [12] Paul M, Mehr AP, Kreutz R. Physiology of local renin-angiotensin systems. Physiol Rev 2006; 86: 747–803.
- [13] Ferreira AJ, Oliveira TL, Castro MCM. Isoproterenol-induced impairment of heart function and remodeling are attenuated by the nonpeptide angiotensin-(1-7) analogue AVE 0991. Life Sci 2007; 81: 916–923.
- [14] Doulton TW, He FJ, MacGregor GA, et al. Systematic review of combined angiotensin-converting enzyme inhibition and angiotensin receptor blockade in hypertension. Hypertension 2005; 45: 880–886.
- [15] Fraga-Sylva RA, Pinheiro SV, GoncŁalves AC, et al. The antithrombotic effect of angiotensin (1-7) involves mas-mediated NO release from platelets. Mol Med 2008; 14: 28–35.
- [16] Yashimoto T, Hirata Y. Aldosterone as a cardiovascular risk hormone. Endocr J 2007; 54: 359–370.
- [17] Shah NC Pringle S, Struthers A. Aldosterone blockade over and above ACE-inhibitors in patients with coronary artery disease but without heart failure. J Renin Angiotensin Aldosterone Syst 2006; 7: 20–30.
- [18] Moreau ME, Garbacki N, Molinaro G, et al. The kallikrein-kinin system: current and future pharmacological targets. J Pharmacol Sci 2005; 99: 6–38.
- [19] Frank MM. Hereditary angioedema: a current state-of-the-art review, VI: novel therapies for hereditary angioedema. Ann Allergy Asthma Immunol 2008; 100 (Suppl. 2): S23–S29.
- [20] What is what. 7 ed., Excerpta Medica, Sweden 2006.
- [21] Sica DA, et al. Pharmacologic treatment of hypertension. in EM Antman: Cardiovascular Therapeutics, Saunders 2007.
- [22] Krum H, Gilbert R, Menard J. Novel therapies blocking the renin-angiotensin aldosterone system in the management of hypertension and related disorders. J Hypertens 2007; 25: 25–35.
- [23] Ferro A, Gilbert R, Krum H. Importance of renin in blood pressure regulation. Int J Clin Pract 2006; 60: 577–581.
- [24] Emmos GT, Argenti R, Martin LL, et al. Pharmacokinetics of M100240 and MDL 100, 173, a dual angiotensin-converting enzyme/neutral endopeptidase inhibitor, in healthy young and elderly volunteers. J Clin Pharmacol 2004; 44: 901–905.
- [25] Seccia TM, Belloni AS, Guidolin D, et al. The renal antifibrotic effects of angiotensin-converting enzyme inhibition involve bradykinin B2 receptor activation in angiotensin II-dependent hypertension. J Hypertens 2006; 24: 1419–1427.
- [26] Jia Y, Lee LY. Role of TRPV receptors in respiratory diseases. Biochim Biophys Acta 2007; 1772: 915–927.
- [27] Ignjatovic T, Tan F, Brovkovych V, et al. Activation of bradykinin B1 receptor by ACE inhibitors. Int Immunopharmacol 2002; 2: 1787–1793.
- [28] Dicpinigaitis PV. Angiotensin-converting enzyme inhibitor-induced cough: ACCP evidence-based clinical practice guidelines. Chest 2006; 129 (1 Suppl.): 169S–173S.
- [29] Pool JL. Direct renin inhibition: focus on aliskiren. J Manag Care Pharm 2007; 13 (8 Suppl B): 21–33.
- [30] Frampton JE, Curran MP. Aliskiren: a review of its use in the management of hypertension. Drugs 2007; 67: 1767–1792.
- [31] Gradman AH, Pinto R, Kad R. Current concepts: renin inhibition in the treatment of hypertension. Curr Opin Pharmacol 2008; 8: 120–126.