Turnerův syndrom v dětství a v adolescenci – současné možnosti léčby
Turnerův syndrom (TS) patří k nejčastějším chromozomálním odchylkám. Postihuje přibližně jedno ze 2000–2500 živě narozených děvčátek. Příčinou TS je deficit jednoho chromozomu X (45,X) nebo jeho části (např. 46,X,i[Xq]), který může postihovat všechny buňky nebo se vyskytuje ve formě chromozomální mozaiky (např. 45,X/46,XX). Vzácně bývá přítomen chromozom Y nebo jeho části (45,X/46,XY). Klasické klinické příznaky zahrnují závažnou poruchu vzrůstu, gonadální dysgenezi, vrozené vady kardiovaskulárního a uropoetického systému, autoimunitní onemocnění, vady smyslových orgánů a možné jiné fenotypové odchylky. Dívky a ženy s TS mají oproti běžné populaci vyšší morbiditu a předčasnou mortalitu, proto musí být od raného věku systematicky sledovány. V dětství je léčena především růstová porucha, v adolescenci se soustřeďuje péče na správné vedení hormonální substituční léčby hypergonadotropního hypogonadismu. Po stanovení diagnózy je nutné vyloučit vrozené vady vnitřních orgánů (zejména srdce a velkých cév) a ostatní přidružené choroby. Dospělé pacientky bez významného kardiovaskulárního rizika mohou využít metod asistované reprodukce. Péče v dospělosti má zejména preventivní charakter vzhledem ke zvýšenému riziku vzniku osteoporózy, dyslipidemie, obezity, diabetu mellitu, hypertenze a dilatace aorty.
Úvod
Turnerův syndrom (TS) je jednou z nejčastějších chromozomálních aneuploidií s incidencí 1 : 2000 až 1 : 2500 živě narozených děvčátek. V České republice tedy žije v současnosti asi 2000 dívek a žen s TS a každoročně přibývá přibližně dalších dvacet. V povědomí lékařů je TS spojen především s růstovou poruchou a gonadální dysgenezí, ale jeho nositelky mohou mít celou řadu dalších zdravotních problémů. Vyhledávají tedy pomoc zdravotníků častěji než ostatní. Téměř každý lékař se během své praxe s pacientkou s TS setká a měl by jí poskytnout péči na základě moderních medicínských poznatků. Nejčastějším typem chromozomální odchylky je u TS chybění jednoho chromozomu X (45,X). Příčinou ztráty je nondisjunkce během meiotického nebo mitotického (postzygotického) dělení. Strukturální abnormity chromozomu X jsou méně časté a vznikají v důsledku chromozomálních zlomů při meióze – např. izochromozom z dlouhých ramének iXq (46,X,i(Xq) nebo ring chromozom rX (46,X,r(X), případně může chybět dlouhé (Xq-) nebo krátké (Xp-) raménko chromozomu X (46,X,del(Xq), respektive 46,X,del(Xp). Tyto abnormity mohou postihovat všechny buňky nebo jen jejich část ve formě chromozomální mozaiky (např. 45,X/46,XX nebo 46,XiXq/45,X). Výjimečně může být přítomen případně chromozom Y (45,X/46,XY) nebo jeho část. Díky rozvoji nových cytogenetických a molekulárních metod jsou původní genetické nálezy přehodnocovány a často jsou odhaleny i 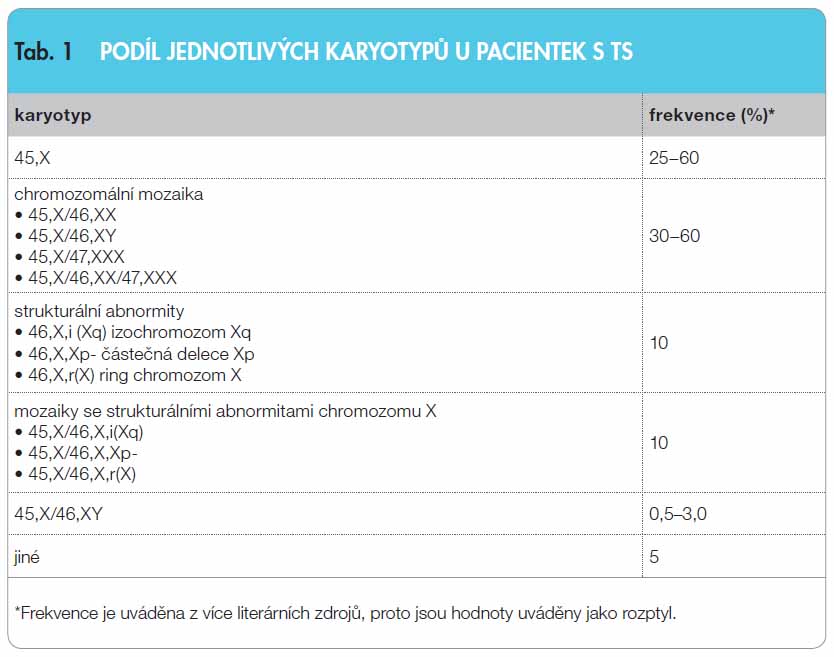 minoritní mozaikové formy TS. Je ověřeno, že abnormity chromozomu X typické pro TS se vyskytují u 1–3 % všech koncepcí, ale 99 % plodů s karyotypem 45,X je spontánně potraceno již v rané fázi gravidity. Toto zjištění vedlo k předpokladu, že větší „životaschopnost“ mají plody s chromozomální mozaikou, jejichž druhý pohlavní chromozom je v některých tkáních alespoň ve zbytku zachován. Někteří autoři udávají, že čistá monozomie (45,X) není slučitelná se životem, a že se tedy u 45,X pacientek jedná vždy o skrytou, nerozpoznanou mozaiku. Přestože nelze tuto hypotézu zatím spolehlivě ověřit, podporují ji nálezy při stanovení karyotypu z buněk více tkání (např. z kožních fibroblastů) nebo při hodnocení velkého počtu mitóz (více než 50) [1]. Zastoupení jednotlivých karyotypů u pacientek s TS je uvedeno v tab. 1.
minoritní mozaikové formy TS. Je ověřeno, že abnormity chromozomu X typické pro TS se vyskytují u 1–3 % všech koncepcí, ale 99 % plodů s karyotypem 45,X je spontánně potraceno již v rané fázi gravidity. Toto zjištění vedlo k předpokladu, že větší „životaschopnost“ mají plody s chromozomální mozaikou, jejichž druhý pohlavní chromozom je v některých tkáních alespoň ve zbytku zachován. Někteří autoři udávají, že čistá monozomie (45,X) není slučitelná se životem, a že se tedy u 45,X pacientek jedná vždy o skrytou, nerozpoznanou mozaiku. Přestože nelze tuto hypotézu zatím spolehlivě ověřit, podporují ji nálezy při stanovení karyotypu z buněk více tkání (např. z kožních fibroblastů) nebo při hodnocení velkého počtu mitóz (více než 50) [1]. Zastoupení jednotlivých karyotypů u pacientek s TS je uvedeno v tab. 1.
Klinický obraz
TS zahrnuje celou řadu klinických příznaků, které nemusí být vždy plně vyjádřeny [2] (tab. 2). Vedoucím symptomem je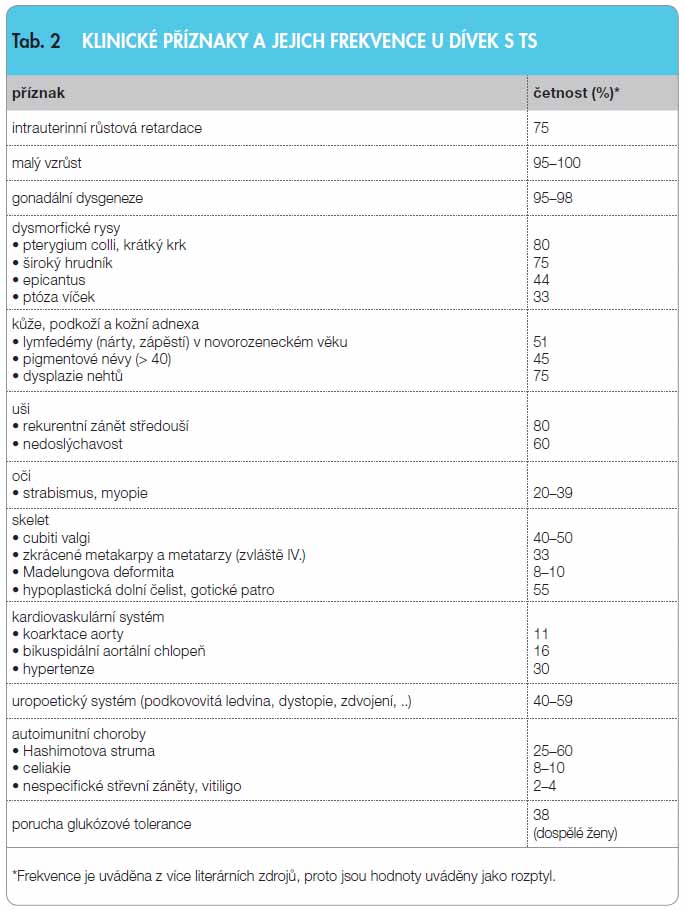 v dětství růstová porucha, proto by u všech dívek, které nerostou v souladu s dědičným růstovým potenciálem, měl být TS vyloučen. Dalšími nejčastějšími příznaky jsou dysgeneze gonád a pozůstatky fetálního lymfedému, mezi něž patří kožní duplikatury po stranách krku (pterygium colli) a nízká vlasová hranice, široký hrudník s oddálenými bradavkami a postnatální lymfedém na dorzech rukou a na nártech. Pro TS je typická vysoká incidence renálních a kardiovaskulárních vad. Nositelky TS nejsou mentálně retardované s výjimkou pacientek s ring chromozomem. S rozmanitou manifestací klinických projevů souvisí věk při stanovení diagnózy TS. Dříve jsou obvykle rozpoznávány pacientky s klasickou formou TS (45,X). Diagnóza mírných forem syndromu, typických pro minoritní mozaiky, může být stanovena až v době adolescence, nebo dokonce v dospělosti, protože tyto pacientky se často svým vzhledem nijak nápadně neodlišují od ostatních děvčat. Přibližně u 50 % děvčat bývají symptomy tak nápadné, že je možno vyslovit podezření na TS již záhy po narození (kongenitální lymfedémy, pterygium colli nebo koarktace aorty), a díky kvalitní prenatální péči (ultrasonografie) přibývá i pacientek diagnostikovaných před narozením. Fenotyp do jisté míry koreluje s karyotypem, resp. genotypem, i když rozdílnost pozorujeme i u pacientek se stejnou chromozomální abnormalitou (obr. 1).
v dětství růstová porucha, proto by u všech dívek, které nerostou v souladu s dědičným růstovým potenciálem, měl být TS vyloučen. Dalšími nejčastějšími příznaky jsou dysgeneze gonád a pozůstatky fetálního lymfedému, mezi něž patří kožní duplikatury po stranách krku (pterygium colli) a nízká vlasová hranice, široký hrudník s oddálenými bradavkami a postnatální lymfedém na dorzech rukou a na nártech. Pro TS je typická vysoká incidence renálních a kardiovaskulárních vad. Nositelky TS nejsou mentálně retardované s výjimkou pacientek s ring chromozomem. S rozmanitou manifestací klinických projevů souvisí věk při stanovení diagnózy TS. Dříve jsou obvykle rozpoznávány pacientky s klasickou formou TS (45,X). Diagnóza mírných forem syndromu, typických pro minoritní mozaiky, může být stanovena až v době adolescence, nebo dokonce v dospělosti, protože tyto pacientky se často svým vzhledem nijak nápadně neodlišují od ostatních děvčat. Přibližně u 50 % děvčat bývají symptomy tak nápadné, že je možno vyslovit podezření na TS již záhy po narození (kongenitální lymfedémy, pterygium colli nebo koarktace aorty), a díky kvalitní prenatální péči (ultrasonografie) přibývá i pacientek diagnostikovaných před narozením. Fenotyp do jisté míry koreluje s karyotypem, resp. genotypem, i když rozdílnost pozorujeme i u pacientek se stejnou chromozomální abnormalitou (obr. 1).
Podle pravděpodobné příčiny vzniku lze jednotlivé symptomy rozdělit do čtyř základních skupin, které mají částečně odlišný etiopatogenetický původ [2]:
- • růstová porucha a kostní abnormity,
- • odchylky v utváření měkkých tkání a vnitřních orgánů,
- • gonadální dysgeneze,
- • neurokognitivní dysfunkce.
Hlavní příčinou je ztráta genů na chromozomu X, které u zdravých osob nepodléhají X-inaktivaci a jejichž dvě funkční kopie jsou nutné pro zabezpečení fyziologického vývoje. Jejich haploinsuficience vede u TS k typickým tělesným odchylkám, zejména k růstové poruše, skeletálním abnormitám včetně zhrubělé kostní struktury (mylně v dětství považované za osteoporózu) a vrozeným vadám měkkých tkání a vnitřních orgánů (SHOX gen a lymfogenní gen). Druhým mechanismem vlivu haploinsuficience genů na chromozomu X na fenotyp TS je genomický imprinting, který vychází z parent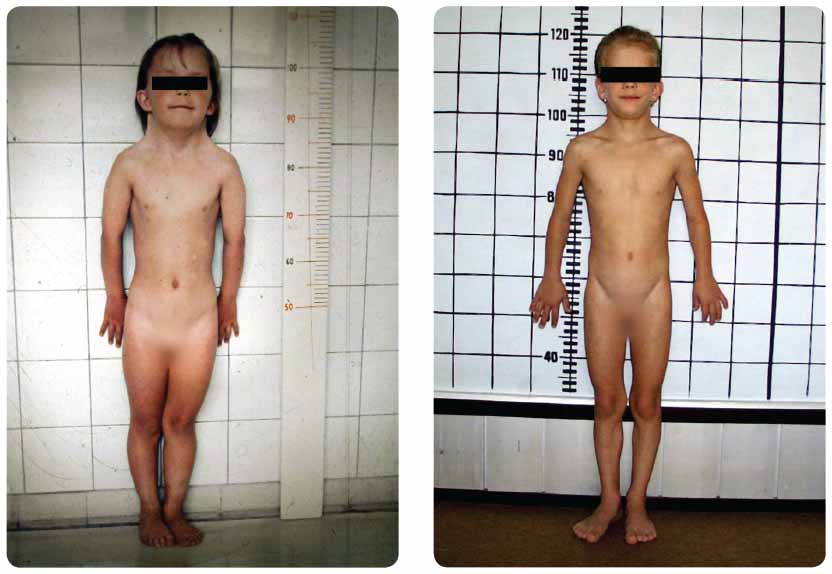 álního původu intaktního chromozomu X. Imprinting má (na rozdíl od genů nepodléhajících X-inaktivaci) vliv především na neurokognitivní fenotyp [3]. Nespecifický chromozomální efekt se uplatňuje především při vzniku a tíži gonadální dysgeneze. Buněčné linie s nepárovými pohlavními chromozomy nevstupují u dívek s TS do meiotického dělení a degenerují. Morfologické změny v ovariích korelují s cytogenetickým nálezem: u chromozomální mozaiky s menším procentuálním zastoupením patologických buněčných linií mohou být ovaria dlouho makroskopicky normální. Postupně ale dochází k fibrotizaci ovariální tkáně, s anovulačními cykly a předčasnou menopauzou [4].
álního původu intaktního chromozomu X. Imprinting má (na rozdíl od genů nepodléhajících X-inaktivaci) vliv především na neurokognitivní fenotyp [3]. Nespecifický chromozomální efekt se uplatňuje především při vzniku a tíži gonadální dysgeneze. Buněčné linie s nepárovými pohlavními chromozomy nevstupují u dívek s TS do meiotického dělení a degenerují. Morfologické změny v ovariích korelují s cytogenetickým nálezem: u chromozomální mozaiky s menším procentuálním zastoupením patologických buněčných linií mohou být ovaria dlouho makroskopicky normální. Postupně ale dochází k fibrotizaci ovariální tkáně, s anovulačními cykly a předčasnou menopauzou [4].
Možnosti léčby Turnerova syndromu
V dětství a adolescenci je zdravotní péče věnována především léčbě závažné růstové poruchy a hypogonadismu. Hledání optimálních léčebných postupů se stalo v 80. letech minulého století předmětem intenzivního výzkumu. Rozhodujícím mezníkem v léčbě růstové poruchy u TS bylo pochopení komplexní příčiny růstové retardace a rozumná koordinace léčby potencující tělesný růst s nutnou estrogenní substitucí.
Růstová porucha a její léčba
Malý vzrůst (disproporcionálního, mezomelického typu) je u TS často prvním symptomem, který vede k diagnóze, a u některých dívek může být znakem jediným. Většina pacientek ho považuje za svůj největší tělesný handicap.
![Graf 1 Percentilový graf tělesné výšky neléčených pacientek s TS ve věku od 2 do 20 let. Tmavá plocha představuje fyziologické rozmezí tělesné výšky zdravých děvčat (upraveno podle [5] – Ranke, et al., 1991).](https://www.remedia.cz/photo-a-28949---.jpg) V růstové retardaci neléčených dívek s TS lze odlišit fázi intrauterinní růstové retardace, která přechází v progredující zaostávání v růstu v období dětství a chybění pubertálního růstového výšvihu v období dospívání. Výsledkem je významně redukovaná adultní výška – v průměru 134–158 cm, tedy o 20 cm méně oproti ženám běžné populace (graf 1) [5]
V růstové retardaci neléčených dívek s TS lze odlišit fázi intrauterinní růstové retardace, která přechází v progredující zaostávání v růstu v období dětství a chybění pubertálního růstového výšvihu v období dospívání. Výsledkem je významně redukovaná adultní výška – v průměru 134–158 cm, tedy o 20 cm méně oproti ženám běžné populace (graf 1) [5]
V minulosti byl za příčinu malého vzrůstu u TS považován hypoestrinní stav. Podávání malých dávek estrogenů v dětském věku bylo založeno na předpokladu, že za fyziologických okolností se estrogeny tvoří i v dětském věku a v pubertě jsou příčinou významného růstového výšvihu. Výsledky byly zpočátku slibné, protože krátkodobě docházelo k urychlení růstové rychlosti, ale paralelně výrazně postupovala kostní zralost, takže docházelo k předčasnému uzávěru epifyzárních štěrbin a dospělá výška pacientek zůstávala v lepším případě nezměněna, obvykle však bývala významně redukována. Od roku 1960 se v literatuře začaly objevovat první zprávy o použití extrakčního růstového hormonu u dívek s TS. Dávkování bylo konvenční – identické s podáváním růstového hormonu pacientům s jeho deficitem. Výsledky léčby substitučními dávkami nebyly povzbudivé. Zahájení výroby rekombinantního růstového hormonu v polovině 80. let umožnilo jeho potenciálně neomezenou výrobu a podnítilo hledání nových indikací. Pozornost se obrátila právě k pacientkám s TS, protože jsou početnou, jednoznačně definovanou skupinou s dobře predikovatelnou růstovou retardací. V tu dobu již bylo potvrzeno, že příčinou malého vzrůstu u TS je haploinsuficience SHOX genu, který se nachází na distální části krátkého raménka chromozomu X (Xp22.33 v PAR 1) a ovlivňuje růst dlouhých kostí, zejména předloktí a bérců [6]. Růstová porucha u TS je tedy formou kostní dysplazie. Tento poznatek vedl k doporučení zvýšit původně substituční dávky růstového hormonu na dávky farmakologické, které jsou přibližně dvojnásobně vyšší (0,05 mg/kg/den) a vedou k suprafyziologickým hladinám inzulinu podobného růstového faktoru I (IGF-I). Ty jsou nepochybně hlavním mediátorem efektu růstového hormonu u TS, ať už představují náhradu váznoucí lokální auto- a parakrinní sekrece IGF-I v růstových chrupavkách, nebo překonávají rezistenci cílových tkání vůči IGF-I [7, 8]. Podávání růstového hormonu patří ve vyspělých zemích ke standardním postupům léčby růstové poruchy u dívek s TS (v České republice od r. 1992). Bylo prokázáno, že dlouhodobá léčba růstovým hormonem zvyšuje růstovou rychlost a tím zlepšuje (případně normalizuje) tělesnou výšku v dospělosti. Výsledky studií ale nejsou jednotné (výškový zisk oproti neléčeným pacientkám se pohybuje v průměru kolem 3,0–16,9 cm) a stále je hledán jednotný konsenzus ve strategii léčby [9, 10]. Bylo opakovaně prokázáno, že nejvýznamnějším faktorem ovlivňujícím dospělou výšku je věk v době zahájení léčby růstovým hormonem a délka trvání léčby. Včasný začátek léčby umožňuje podávat růstový hormon po dostatečně dlouhý časový interval před začátkem indukované nebo spontánní puberty [11]. Estrogenní substituce může být za těchto podmínek zahájena mezi 11. a 12. rokem bez rizika předčasného uzávěru růstových štěrbin. U dívek, které neměly možnost být léčeny růstovým hormonem před 11. rokem věku, se doporučuje postupovat individuálně a léčbu estrogeny odložit. Diskutovaným problémem je i hledání optimální dolní věkové hranice pro zahájení léčby růstovým hormonem. Předběžné výsledky randomizovaných studií z posledních 5 let považují za bezpečné začít s léčbou již před 4. rokem věku [12], což vede k nižším finančním nákladům na léčbu, k eliminaci handicapu z malé výšky ještě před zahájením školní docházky a k přiměřenému věku zahájení estrogenní substituce. Předpokladem je samozřejmě včasná diagnostika TS. Některá pracoviště (především v USA) doporučují podpořit růstové tempo u dívek starších 9 let současným podáváním anabolického steroidu oxandrolonu [12, 13]. Tento systém léčby ale nebyl všeobecně akceptován. Léčba růstovým hormonem se ukázala být bezpečná, jak bylo prokázáno retrospektivním hodnocením početných skupin dívek s TS [13].
V České republice je léčba růstovým hormonem soustředěna do specializovaných center. V roce 2010 byla analyzována data od 165 pacientek s TS, které byly léčeny alespoň 12 měsíců růstovým hormonem v období 1992–2009 v devíti centrech a dosáhly své finální výšky. Medián věku diagnózy TS byl 10,0 (0,1–16,4) let, léčba růstovým hormonem byla u nich zahájena ve věku 12,1 (3,5–16,5) let. Léčeny byly standardní denní dávkou 0,050 mg/kg. Spontánní dospívání mělo 22,5 % dívek ve věku 12,0 (8,9–16,0) let, věk na počátku indukované puberty byl 13,9 (8,5–14,8) let. Medián dospělé výšky byl 155,0 (137,5–168,8) cm. Výškový zisk tedy představoval 9 cm. Dívky se spontánní pubertou dosáhly výšky 153,0 (143,0–162,8) cm, pacientky s indukovanou pubertou 156,0 (137,5–168,8) cm. Významným faktorem ovlivňujícím dosaženou finální výšku byl věk zahájení léčby růstovým hormonem, věk počátku puberty a doba podávání růstového hormonu bez současného podávání estrogenů. Průměrný věk diagnózy TS se snížil z 10,5 ± 4,7 roku (pacientky narozené v letech 1974–1984) na 8,2 ± 4,9 roku (pacientky narozené po r. 1984).
Gonadální dysgeneze, hormonální substituční léčba
Ovariální nedostatečnost (úplná nebo částečná) je přítomna u většiny pacientek s TS a je po růstové poruše druhým nejčastějším symptomem. U 20–30 % pacientek je alespoň částečně zachována sekrece estrogenů dostačující ke spontánnímu vývoji prsních žláz, ale pouze u poloviny z nich je natolik vydatná, aby vyvolala menstruaci, a jen výjimečně bývá pozorována dlouhodobě dostatečná činnost ovarií umožňující pravidelný menstruační cyklus a fertilitu (2–5 %). Obvykle se jedná o pacientky s chromozomální mozaikou s vyšším procentem fyziologických buněčných linií 46,XX, které mají částečně zachovanou normální ovariální tkáň. Postupně ale i u většiny z nich dochází k anovulačním cyklům a k předčasné menopauze [14].
Podmínkou zahájení hormonální substituční léčby (HRT) je potvrzení ovariální insuficience. Léčba je indikována u pacientek bez vlastní dostatečné tvorby pohlavních hormonů. Klasickým projevem je vysoká hladina gonadotropinů – folikulostimulačního (FSH) a luteinizačního hormonu (LH). Tento tzv. hypergonadotropní hypogonadismus lze u děvčátek s TS predikovat již časně postnatálně. Sekrece gonadotropinů (především FSH) má bifázický průběh a první několikanásobný vzestup proti normě je patrný již v novorozeneckém období a je ukazatelem budoucí ovariální nedostatečnosti. V době očekávaného počátku puberty dosahují gonadotropiny až postmenopauzálních hodnot (FSH > 60 mIU/l).
Smyslem estrogenně-gestagenní substituce je u pacientek s TS napodobit přirozenou hormonální produkci zdravých dívek a žen ve stejném biologickém věku. Substituce umožňuje přiměřený vývoj druhotných pohlavních znaků, růst dělohy pro případnou asistovanou reprodukci, ovlivňuje pozitivně nárůst kostní hmoty, kardiovaskulární funkce, jaterní enzymy, vývoj mozku a jiné estrogen-dependentní procesy. Přestože se optimální strategie léčby při indukci puberty stále diskutuje, byly stanoveny doporučené postupy pro podávání estrogenní substituce k navození postupné pohlavní zralosti a její udržení až do období klimakteria [13, 15].
Substituční podávání estrogenů u dívek s Turnerovým syndromem má fázi indukční (monoterapie – od první dávky 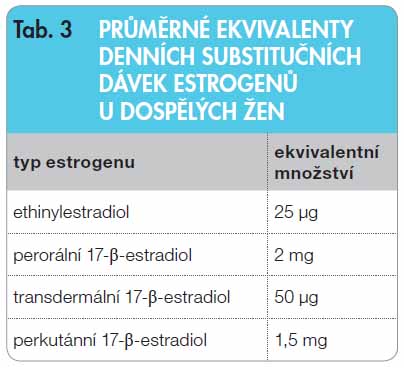 estrogenu po menarché) a fázi udržovací (cyklická estrogen-gestagenní léčba od menarché do postmenopauzy). Věk počátku HRT je nutné posuzovat individuálně s ohledem na výšku dívky a její růstovou prognózu. I velmi malé dávky estrogenů předčasně podané urychlují významně kostní zrání a tím negativně ovlivňují konečnou výšku. Za optimální považuje většina autorů věk 12–13 let a jen při velmi špatné růstové prognóze doporučuje léčbu zahajovat až po 14. roce. Doporučená počáteční dávka estrogenů by měla odpovídat asi jedné desetině substituční dávky dospělých žen (tj. 0,2 mg perorálního 17-beta-estradiolu nebo 2,5 µg ethinylestradiolu nebo 5 µg transdermálního 17-beta-estradiolu). Průměrné ekvivalenty denních substitučních dávek estrogenů uvádí tab. 3. Během indukční fáze trvající 2–3 roky se dávka estrogenu ve 3–6měsíčních intervalech postupně zvyšuje až na dávku adultní (tab. 4), při které se může dostavit první odlučovací krvácení. Toto krvácení je dysfunkční a nelze je považovat za přirozené menarché, protože děložní sliznice není bez p
estrogenu po menarché) a fázi udržovací (cyklická estrogen-gestagenní léčba od menarché do postmenopauzy). Věk počátku HRT je nutné posuzovat individuálně s ohledem na výšku dívky a její růstovou prognózu. I velmi malé dávky estrogenů předčasně podané urychlují významně kostní zrání a tím negativně ovlivňují konečnou výšku. Za optimální považuje většina autorů věk 12–13 let a jen při velmi špatné růstové prognóze doporučuje léčbu zahajovat až po 14. roce. Doporučená počáteční dávka estrogenů by měla odpovídat asi jedné desetině substituční dávky dospělých žen (tj. 0,2 mg perorálního 17-beta-estradiolu nebo 2,5 µg ethinylestradiolu nebo 5 µg transdermálního 17-beta-estradiolu). Průměrné ekvivalenty denních substitučních dávek estrogenů uvádí tab. 3. Během indukční fáze trvající 2–3 roky se dávka estrogenu ve 3–6měsíčních intervalech postupně zvyšuje až na dávku adultní (tab. 4), při které se může dostavit první odlučovací krvácení. Toto krvácení je dysfunkční a nelze je považovat za přirozené menarché, protože děložní sliznice není bez p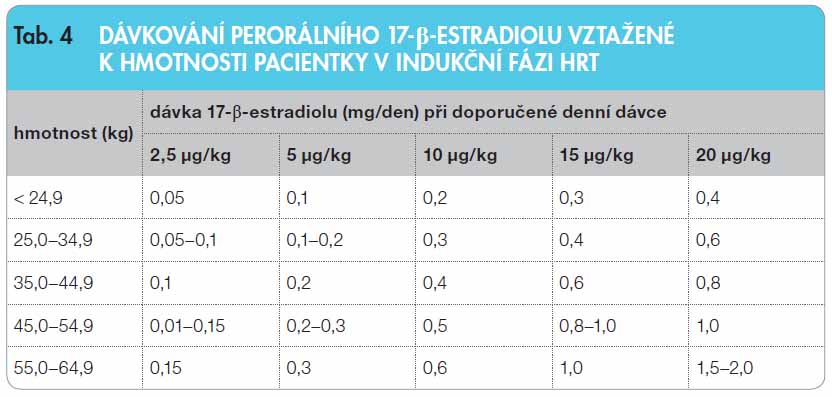 odaného gestagenu sekrečně transformovaná. Fyziologičtější variantou je indukovat první menstruaci přidáním gestagenu v době, kdy je dle ultrasonografického vyšetření (USG) výška děložní sliznice vyšší než 4 mm, což zpravidla odpovídá denní dávce 17-beta- -estradiolu 1,5–2 mg, gestagen – nejčastěji medroxyprogesteron-acetát – v dávce 5–10 mg se přidává na 10–14 dnů v druhé polovině cyklu.
odaného gestagenu sekrečně transformovaná. Fyziologičtější variantou je indukovat první menstruaci přidáním gestagenu v době, kdy je dle ultrasonografického vyšetření (USG) výška děložní sliznice vyšší než 4 mm, což zpravidla odpovídá denní dávce 17-beta- -estradiolu 1,5–2 mg, gestagen – nejčastěji medroxyprogesteron-acetát – v dávce 5–10 mg se přidává na 10–14 dnů v druhé polovině cyklu.
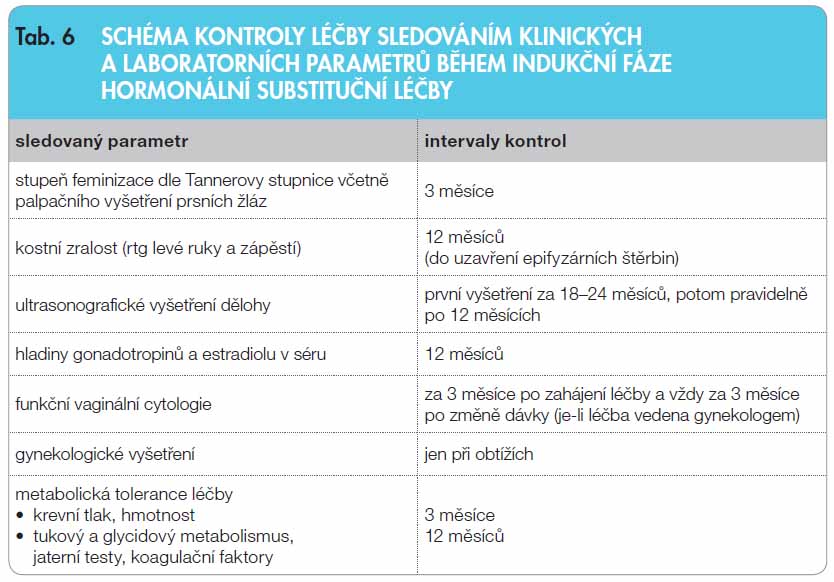
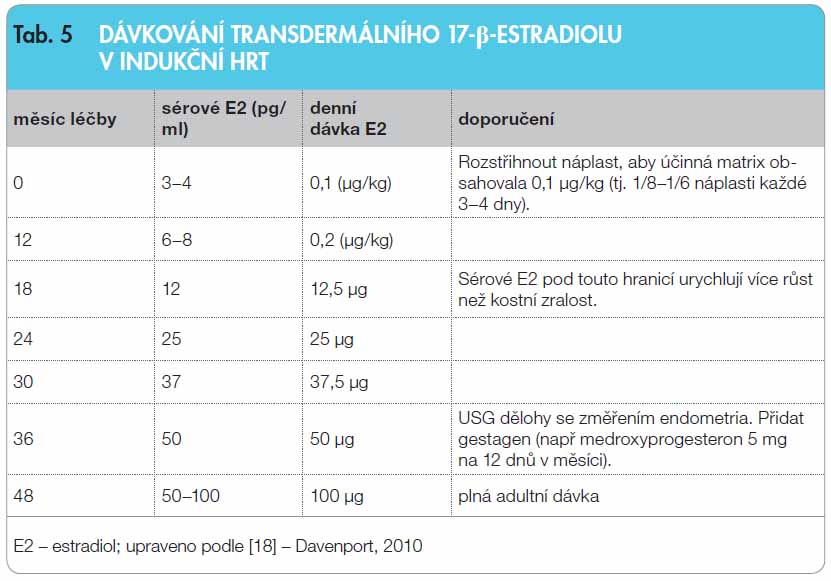 Nejčastěji užívaným estrogenem je perorální 17-beta-estradiol, ale stále častěji se užívají transdermální preparáty (náplasti nebo gel); nejsou metabolizovány v játrech [16], což je jejich výhodou, ale v době rané indukční fáze je problematické jejich dávkování (tab. 5). Vývoj sekundárních pohlavních znaků je nutné sledovat v intervalu 3–6 měsíců. V ročních intervalech je hodnocen stupeň kostní zralosti. Růst dělohy je kontrolován pomocí USG. Hladiny gonadotropinů vykazují sice při stoupající dávce estrogenů postupný pokles, ale prakticky nikdy nedochází k jejich normalizaci. Na strategii léčby nemá hodnocení jejich sérových hodnot rozhodující vliv. Ke zjištění stupně estrogenizace je gynekology doporučováno využít funkční cytologie poševní sliznice (FC), jež může být vodítkem při vedení estrogenní substituce. Schéma kontroly léčby sledováním klinických a laboratorních parametrů během indukční fáze HRT uvádí tab. 6.
Nejčastěji užívaným estrogenem je perorální 17-beta-estradiol, ale stále častěji se užívají transdermální preparáty (náplasti nebo gel); nejsou metabolizovány v játrech [16], což je jejich výhodou, ale v době rané indukční fáze je problematické jejich dávkování (tab. 5). Vývoj sekundárních pohlavních znaků je nutné sledovat v intervalu 3–6 měsíců. V ročních intervalech je hodnocen stupeň kostní zralosti. Růst dělohy je kontrolován pomocí USG. Hladiny gonadotropinů vykazují sice při stoupající dávce estrogenů postupný pokles, ale prakticky nikdy nedochází k jejich normalizaci. Na strategii léčby nemá hodnocení jejich sérových hodnot rozhodující vliv. Ke zjištění stupně estrogenizace je gynekology doporučováno využít funkční cytologie poševní sliznice (FC), jež může být vodítkem při vedení estrogenní substituce. Schéma kontroly léčby sledováním klinických a laboratorních parametrů během indukční fáze HRT uvádí tab. 6.
 Po prvním krvácení se automaticky přechází na sekvenční hormonální substituční léčbu, jejímž cílem je zajistit pravidelný menstruační cyklus a navodit v hlavních cílových orgánech (děloze, poševní sliznici, prsní žláze) typické změny, které jej za fyziologických poměrů provázejí. Cyklická léčba estrogeny a gestageny umožňuje udržet přiměřený rozvoj sekundárních pohlavních znaků, přispívá k uspokojivému pohlavnímu životu a plné identifikaci s ženskou rolí. Optimální vývoj dělohy je předpokladem úspěchu pro případnou léčbu neplodnosti metodami asistované reprodukce s využitím darovaného oocytu. Dlouhodobá substituční léčba slouží rovněž k prevenci osteoporózy a kardiovaskulárních změn. Během cyklické HRT jsou další preventivní gynekologické kontroly indikovány podobně jako u zdravé (nerizikové) ženské populace 1krát za rok, ale navíc je doporučeno sledování metabolické tolerance HRT (krevní tlak, hmotnost, tukový a sacharidový
Po prvním krvácení se automaticky přechází na sekvenční hormonální substituční léčbu, jejímž cílem je zajistit pravidelný menstruační cyklus a navodit v hlavních cílových orgánech (děloze, poševní sliznici, prsní žláze) typické změny, které jej za fyziologických poměrů provázejí. Cyklická léčba estrogeny a gestageny umožňuje udržet přiměřený rozvoj sekundárních pohlavních znaků, přispívá k uspokojivému pohlavnímu životu a plné identifikaci s ženskou rolí. Optimální vývoj dělohy je předpokladem úspěchu pro případnou léčbu neplodnosti metodami asistované reprodukce s využitím darovaného oocytu. Dlouhodobá substituční léčba slouží rovněž k prevenci osteoporózy a kardiovaskulárních změn. Během cyklické HRT jsou další preventivní gynekologické kontroly indikovány podobně jako u zdravé (nerizikové) ženské populace 1krát za rok, ale navíc je doporučeno sledování metabolické tolerance HRT (krevní tlak, hmotnost, tukový a sacharidový 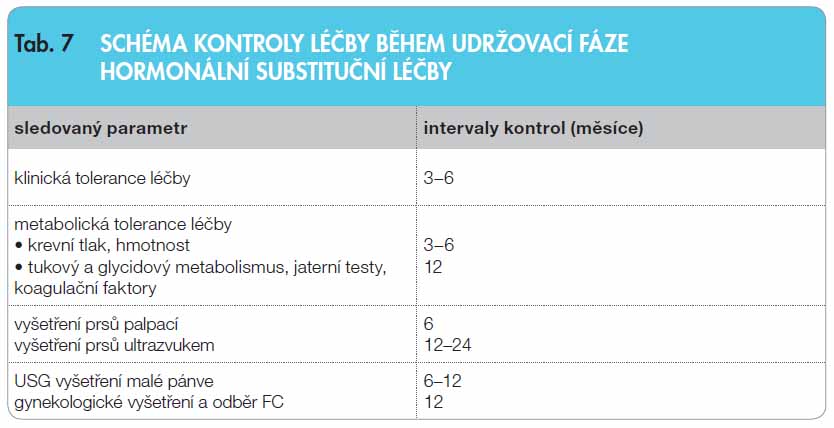 metabolismus, jaterní testy, koagulační faktory). Schéma kontrol během léčby uvádí tab. 7. Pacientky s TS mají často zvýšené hladiny jaterních enzymů (AST, ALT, GMT, ALP), zatímco hodnoty bilirubinu a koagulační parametry se nacházejí ve fyziologických rozmezích. Za příčinu hepatopatie jsou považovány vrozené vaskulární změny a non-alkoholická jaterní steatóza. Podání HRT jaterní funkce nezhoršuje, jak bylo prokázáno v četných studiích [17], a není důvodem k nezahájení HRT nebo k jejímu přerušení!
metabolismus, jaterní testy, koagulační faktory). Schéma kontrol během léčby uvádí tab. 7. Pacientky s TS mají často zvýšené hladiny jaterních enzymů (AST, ALT, GMT, ALP), zatímco hodnoty bilirubinu a koagulační parametry se nacházejí ve fyziologických rozmezích. Za příčinu hepatopatie jsou považovány vrozené vaskulární změny a non-alkoholická jaterní steatóza. Podání HRT jaterní funkce nezhoršuje, jak bylo prokázáno v četných studiích [17], a není důvodem k nezahájení HRT nebo k jejímu přerušení!
Závěr
Přestože většina postupů nevychází z medicíny založené na důkazech, ale od skupin vědců a kliniků, kteří mají dlouholetou medicínskou zkušenost s pacientkami s TS, došlo v posledních dvaceti letech ve vyspělých zemích k významnému pokroku v diagnostice a léčbě dívek s TS [18]. Uznávaným mezníkem byla 90. léta minulého století, kdy se péče o pacientky s TS soustředila do center dětské endokrinologie. Významně se snížila věková hranice stanovení diagnózy TS, což umožňuje včasné zahájení léčby růstovým hormonem a tím zahájení estrogenní substituce v přiměřeném věku. Zlepšila se diagnostika kardiovaskulárních odchylek, které jsou nejčastější příčinou morbidity a časné mortality [19]. Právě období dětství a dospívání je rozhodující – jen včasná diagnóza a odpovídající léčba umožňují zlepšit kvalitu jejich života i v dospělosti.
Seznam použité literatury
- [1] Jacobs P, Dalton P, James R, et al. Turner syndrome: a cytogenetic and molecular study. Ann Hum Genet 1997; 6: 471–483.
- [2] Zapletalová J, Lebl J. Klinická symptomatologie Turnerova syndromu. In: Zapletalová J, Lebl J, Šnajderova M. (Eds), Turnerův syndrom. Praha: Galén, 2003: 68–109.
- [3] Bondy CA. Genomic imprinting in Turner syndrome. In: Gravholt, CH, Bondy CA (Eds), Welness for Girls and Women with Turner syndrome. Amsterdam: Elsevier, BV, 2006: 21–25.
- [4] Modi DN, Sane S, Bhartiya D. Accelerated germ cell apoptosis in sex chromosome aneuploid fetal human gonads. Mol Hum Reprod 2003; 9: 219–225.
- [5] Ranke MB, Chavez-Mayer H, Blank B, et al. Spontaneous growth and bone age development in Turner syndrome: Results of a multicentric study 1990. In: Ranke MB, Rosenfeld RG, eds. Turner syndrome: Growth promoting therapies. Amsterdam, New York, Oxford: Excerpta Medica, 1991: 101–112.
- [6] Rao E, Weiss B, Fukami M, et al. Pseudoautosomal deletions encompassing a novel homeobox gene cause growth failure in idiopathic short stature and Turner syndrome. Nat Genet 1997; 16: 54–63.
- [7] Hochberg Z, Aviram M, Rubin D, Pollack S. Decreased sensitivity to insulin-like growth factor I in Turner´s syndrome: a study of monocytes and T lymphocytes. Eur J Clin Invest 1997; 27: 543–547.
- [8] Westwood M, Tajbakhsh SH, Siddals KW, et al. Reduced pericellular sensitivity to IGF-I in fibroblasts from girls with Turner syndrome: a mechanism to impair clinical responses to GH. Pediatr Res 2011; 70: 25–30.
- [9] Cave CB, Bryant J, Milne R. Recombinant growth hormone in children and adolescents with Turner syndrome. Cochrane Database Syst Rev 2003; 1: CD003887.
- [10] Stephure DK, Canadian Growth Hormone Advisory Committee. Impact of growth hormone supplementation on adult height in Turner syndrome: results of the Canadian randomized controlled trial. J Clin Endocrinol Metab 2005; 90: 3360–3366.
- [11] Bondy CA, Turner Syndrome Study Group. Care of girls and women with Turner syndrome: a guideline of the Turner Syndrome Study Group. J Clin Endocrinol Metab 2007; 92: 10–25.
- [12] Davenport ML, Crowe BJ, Travers SH, et al. Growth hormone treatment of early growth failure in toddlers with Turner syndrome: a randomized, controlled, multicenter trial. J Clin Endocrinol Metab 2007; 92: 3406–3416.
- [13] Rosenfeld RG. Turner syndrome – growth hormone treatment. In: Ranke MB, Price DA, Reiter EO. (Eds), Growth hormone therapy in pediatrics – Literatura 20 years of KIGS. Basel, Karger 2007: 326–331.
- [14] Pasquino AM, Passeri F, Pucarelli I, et al. Spontaneous pubertal development in Turner‘syndrome. Italian Study Group for Turner‘s Syndrome. J Clin Endocrinol Metab 1997; 82: 1810–1813.
- [15] Kiess W, Conway G, Ritzen M, et al. Induction of puberty in the hypogonadal girls – practices and attitudes of pediatric endocrinologist in Europe. Horm Res 2002; 57: 66–71.
- [16] Nabhan ZM, DiMeglio LA, Qi R, et al. Conjugated oral versus transdermal estrogen replacement in girls with Turner syndrome: a pilot comparative study. J Clin Endocrinol Metab 2009; 94: 2009–2014.
- [17] Gravhold CH, Poulsen HE, Ott P, et al. Quantitative liver functions in Turner syndrome with and without hormone replacement therapy. Eur J Endocrinol 2007; 156: 679–686.
- [18] Davenport ML. Approach to the patient with Turner syndrome. J Clin Endocrinol Metab 2010; 95: 1487–1495.
- [19] Stochholm K, Juul S, Juel K, et al. Prevalence, incidence, diagnostic delay, and mortality in Turner syndrome. J Clin Endocrinol Metab 2006; 91: 3897–3902.